Definizione dell’Omocisteina
L’omocisteina è un aminoacido non proteico prodotto dal metabolismo della metionina, un’aminoacido solforato essenziale che viene introdotto nel nostro organismo con la dieta.
Metabolismo dell’Omocisteina
Il metabolismo dell’omocisteina può seguire due vie (Fig.1):
via della rimetilazione: utilizza gli enzimi metionina-sintasi, metilenetetraidrofolatoreduttasi (MTHFR), betaina-sintasi e ha come catabolita finale la metionina. In tale via l’omocisteina può essere ri-metilata a metionina mediante due processi. Nel primo, in cui è fondamentale l’acido folico, la reazione chiave avviene grazie all’enzima MTHFR che riduce il 5,10-metilene-tetraidrofolato a 5-metiltetraidrofolato; quest’ultimo fornirà poi, in presenza di un coenzima, la vitamina B12, il gruppo metilico necessario per la riconversione dell’omocisteina in metionina. Nel secondo processo invece la reazione di rimetilazione è svolta dall’enzima betaina-sintasi che produce metionina catalizzando il trasferimento di un gruppo metilico dalla betaina all’omocisteina.
via della transulfurazione: sfrutta l’enzima cistationina--sintasi e ha come prodotto finale l’aminoacido cisteina. La cistationina--sintasi, coadiuvata dal coenzima vitamina B6, catalizza la reazione di condensazione tra omocisteina e serina con formazione di cistationina che successivamente viene degradata a cisteina.
Mentre la via metabolica della rimetilazione è attiva per basse concentrazioni di omocisteina e di metionina, la via della transulfurazione entra in gioco quando le concentrazioni dei due aminoacidi aumentano. Inoltre, si pensa che la transulfurazione dell’omocisteina e la sua rimetilazione-betaina dipendente avvengano esclusivamente nel fegato, e che la via della rimetilazione folato/vitamina B12 dipendente sia l’unica trasformazione metabolica dell’omocisteina operante nei distretti cellulari periferici. Quando si ha la saturazione delle vie metaboliche, l’omocisteina intracellulare in eccesso viene esportata nella circolazione (aumentando così i livelli plasmatici di omocisteina) e lì si lega alle proteine plasmatiche oppure viene eliminata, principalmente dal rene.
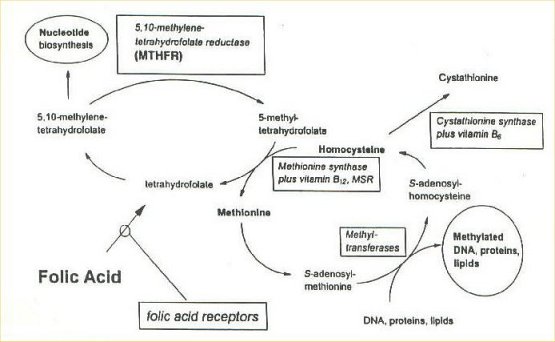
Fig.1 Metabolismo dell’Omocisteina
Iperomocisteinemia
Con il termine iperomocisteinemia, si intende la presenza di elevati livelli di omocisteina nel sangue. In Italia, la maggior parte dei laboratori considera normali per gli adulti concentrazioni di omocisteina inferiori a 13 μmol/l per gli uomini e inferiori a 10.1 μmol/l per le donne; per i bambini fino ai 14 anni sono considerate normali concentrazioni inferiori a 11.3 μmol/l.
L’iperomocisteinemia è attualmente considerata un importante fattore di rischio per lo sviluppo di malattie cardiovascolari (aterosclerosi coronarica ed infarto miocardico), cerebrovascolari (ictus cerebrale) e vascolari periferiche (trombosi arteriose e venose).
Si stima che le persone con iperomocisteinemia abbiano un rischio circa doppio di sviluppare una malattia cardiovascolare rispetto a chi ha dei valori normali.
Elevati livelli ematici di omocisteina si riscontrano inoltre in alcune malattie (ipotiroidismo, psoriasi, lupus eritematoso sistemico, artrite reumatoide) e durante trattamenti con alcuni farmaci (metotrexate, carbamazepina, fenitoina ed isoniazide).
Cause di Iperomocisteinemia
La concentrazione plasmatica di omocisteina è il risultato di una stretta relazione tra le abitudini dietetiche e i fattori genetici predisponenti.
E’ possibile riscontrare elevati livelli di omocisteina nel sangue a causa di una dieta non sufficientemente ricca di acido folico e delle altre vitamine del gruppo B.
L’iperomocisteinemia risulta dunque essere causata da un insieme di più fattori: molti di questi possono essere difficilmente modificati (stati patologici, terapie o condizioni acquisite,...) mentre altri, legati a determinati stili di vita, sono invece modificabili (tabagismo, eccessivo consumo di caffè e di bevande alcoliche, alimentazione non equilibrata, ridotta attività fisica,...). Pertanto l’astensione dal fumo, la riduzione del consumo di caffè e di bevande alcoliche, variazioni nella dieta e supplementazione vitaminica (specialmente Vit. B6, B12, e folati) possono ridurre i livelli di omocisteina anche in presenza di altre cause.
Valori elevati di omocisteina (10-50 volte) si hanno in errori congeniti del metabolismo allo stato omozigote ed in grado minore allo stato eterozigote di:
1. Deficit di cistationin-beta- sintasi (CBS) il cui gene è posto sul braccio lungo del cromosoma 21: sino ad oggi sono stati riportati 17 tipi di mutazione del gene della CBS. Trasmissione autosomica recessiva. Allo stato omozigote prevalenza 1: 200.000, allo stato eterozigote 0.3-1.5% della popolazione. I valori di omocisteina sono molto più elevati nello stato omozigote rispetto allo stato eterozigote, dove risultano per altro sempre aterogeni. Nello stato omozigote si ha il quadro dell’omocistinuria, perché l’omocisteina si trasforma in omocistina che viene eliminata per via urinaria ed essendo poco solubile porta spesso a litiasi urinaria radiopaca. In questi casi sono presenti complicanze tromboemboliche in giovane età, con un rischio superiore al 50 % a partire dai 30 anni di età. Tali complicanze possono portare ad ictus, ad infarto miocardico, ad ipertensione renovascolare, a claudicatio intermittens, ad ischemia mesenterica, ad embolia polmonare. E’ presente una aterosclerosi prococe diffusa, comportante elevata morbilità e mortalità.
2. Deficit di 5-10-metilentetraidrofolatoreduttasi (MTHFR), il cui gene è sito sul braccio corto del cromosoma 1. Molto frequente (prevalenza del genotipo omozigotico dell’ 8-15% della popolazione e fino al 65% della popolazione nel genotipo eterozigotico), trasmissione autosomica recessiva, variabile grado di assenza (80-100%) o variante termolabile dell’enzima. Sono state descritte molte mutazioni di questo gene: la mutazione del gene 667 C—-T comporta sintesi di alanina al posto dell’aminoacido valina. Ciò causa diminuzione dell’attività enzimatica specifica, aumentata termolabilità, elevati valori plasmatici di omocisteina: questa mutazione emerge tuttavia come fattore di rischio cardiovascolare solo in soggetti con basso status di folati. Ciò sottolinea l’importanza, nella prevenzione e nella terapia, dell’apporto nutrizionale di acido folico, il cui deficit risulta un cofattore patogeno necessario.
3. Deficit di metionin-sintetasi, da carenza di vitamina B12.
Omocisteina e danno vascolare
I meccanismi con cui l’omocisteina plasmatica provoca danni a livello vasale non sono ancora del tutto noti.
L’omocisteina risulta essere altamente lesiva per l’endotelio: con il passare del tempo questa istolesività aumenta fino a provocare la trombosi. Inoltre, sembra che l’omocisteina da una parte antagonizzi la sintesi e la funzione dell’ossido nitrico endoteliale, riducendo in tal modo l’azione vasodilatante ad esso legata, e, dall’altra, provochi la formazione dell’anione superossido (O2-) precursore del radicale citotossico perossinitrito, con conseguente aggravamento dello stress ossidativo.
Un possibile meccanismo attraverso il quale l’omocisteina favorisce l’aterosclerosi potrebbe essere quello legato alla proliferazione delle cellule muscolari lisce, che a sua volta determina un aumento dell’adesione endoteliale con un incremento della deposizione di lipoproteine a bassa densità (LDL) e formazione di cellule schiumose. Inoltre l’omocisteina sembrerebbe agire direttamente sull’attivazione piastrinica con un aumento sia dell’adesione che dell’aggregazione.
Il meccanismo attraverso il quale l’omocisteina porta alla formazione di trombi sembra essere legato all’attivazione del fattore V endoteliale, all’inibizione della proteina C e alla riduzione dell’attività dell’antitrombina III.
Iperomocisteinemia come fattore di rischio cardiovascolare: il caso di atleti agonisti
Le malattie cardiovascolari rappresentano la maggiore causa di morbilità e mortalità nei paesi occidentali. I tradizionali fattori di rischio, come l’ipercolesterolemia, l’ipertensione, il diabete ed il fumo, non rendono tuttavia ragione di tutti i casi di queste patologie. Nel campo della cardiopatia coronarica, ad esempio, il solo fattore colesterolo risulta non essere la causa in oltre il 35% dei casi. In alcuni soggetti il solo fattore di rischio evidente è una storia familiare di malattia cardiovascolare precoce, spesso tuttavia senza una chiara predisposizione genetica.
Emerge quindi la necessità di identificare altri marcatori di rischio cardiovascolare che accrescano le conoscenze sui meccanismi fisiopatologici della malattia e che permettano lo sviluppo di nuove misure preventive e terapeutiche.
Come già detto, l’iperomocisteinemia costituisce un fattore di rischio cardiovascolare importante ed indipendente: l’aumento della concentrazione plasmatica a digiuno dell’omocisteina, come dimostrato da numerosi studi clinici, è associato ad un incremento del rischio di infarto miocardico, ictus cerebrale, vasculopatia periferica e trombosi. Negli ultimi vent’anni molto interesse ha dunque destato il possibile ruolo dell’aumento dell’omocisteina nel plasma nella patogenesi della malattia cardiovascolare e tromboembolica.
E’ noto che l’attività fisica è in grado di migliorare i fattori di rischio cardiovascolare quali l’incremento delle lipoproteine e l’ipertensione. La massima funzione preventiva dell’attività fisica, dal punto di vista epidemiologico, è stata dimostrata per le malattie cardiovascolari e metaboliche. Numerose evidenze scientifiche hanno dimostrato che praticare attività fisica può ritardare l’insorgenza di diverse patologie quali il diabete mellito non insulino-dipendente, l’osteoporosi e la sindrome metabolica.
E’ altresì vero che l’attività fisica intensa protratta per lunghi periodi può essere dannosa per la salute dell’atleta. A tal riguardo ben noti sono gli effetti dannosi dello stress psico-fisico derivante dall’attività sportiva agonistica che possono sfociare nella conosciuta sindrome da overtraining. Dati riportati in letteratura hanno evidenziato come l’attività sportiva agonistica aumenti l’incidenza di alcuni stati patologici (es. danno da sovraccarico, exercised induced astma, riduzione dell’immunità, disturbi ormonali, riduzione della fertilità) e sia in grado di rendere manifesti difetti funzionali altresì quiescenti (es. long-QT-syndrome con casi di morte improvvisa).
Negli ultimi anni sono stati condotti alcuni studi per verificare l’esistenza di una correlazione tra attività fisica agonistica e il rischio cardiovascolare. I dati ottenuti sono contrastanti: alcuni lavori indicano una maggiore incidenza del rischio cardiovascolare in atleti d’elite, altri invece riportano una riduzione del rischio in atleti praticanti attività fisica moderata.
Tra i fattori di rischio cardiovascolare indagati, l’omocisteina riveste un ruolo preminente essendo stato evidenziato un suo aumento significativo negli atleti agonisti