FATAL FAMILIAL INSOMNIA: a waking nightmare
DEFINITION
Fatal Familial Insomnia (FFI) is a very rare disease belonging to the family of human transmissible spongiform encephalopathies.
Way of transmission: FFI is a genetic autosomal dominant disease. The point mutation (Met/Val) is transmitted from the parent to the offspring with a probability of 50%.
Pathogenetic mechanism: FFI is due to an alteration of prions, proteins normally present in the human brain that are altered in the patient: a point mutation in the PrP gene causes the synthesis of easily degradable proteins which build up in the thalamus, causing the alteration of areas implicated in the regulation of sleep-wake rhythms. Anyway there's no quantitative association between prions and thalamic degeneration, that is to say that number of modified prions in an area is not directly proportional to damage severity.
In conclusion, two hypothesis about prion role have been elaborated: either the abnormal protein is adverse on neurons or it is not and it leads only to the loss of cellular signals.
Clinical manifestation: Anyone who lives with the problem of insomnia knows how frustrating this condition can be: individuals suffering from FFI manifest initially weakness and classic symptoms of depression; later hours of sleep decrease progressively until they become completely absent. In reality, however, the patient seems to sleep all the time, pouring in a state of confusion that resembles sleepwalking. The course may vary from a few months to two/three years, with unfortunately fatal outcome.
FFI affects about 200 individuals in the world, condemning them to a real curse that takes away sleep and flows disorder into the brain, making it impossible to distinguish the reality from dreams. In Austria, China, Japan and the U.S. some cases have been identified, in Italy, at present, in two families there are people suffering from this disease.
PRION
In FFI a point mutation causes the synthesis of abnormal proteins.
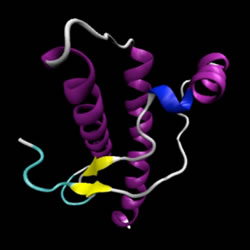
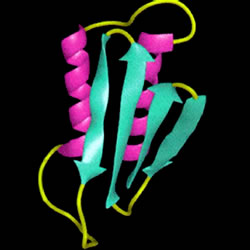
Prion, which stands for "Proteinaceus Infective ONly particle", is the name given by Dr.Stanley B. Prusiner to a hypothetical "unconventional infectious agent" of proteic nature. It is a conformational isomer of a glycoprotein normally expressed on the surface of all nucleated cells, predominantly on neurons. It does not contain nucleic acid and it is therefore resistant to the action of all those enzymes that destroy DNA and RNA. It is also highly resistant to heat and UV rays.
Prions are responsible for a number of diseases in a variety of organisms, including bovine spongiform encephalopathy and fatal familial insomnia in humans. All known prion animal diseases affect the structure of the CNS or other neural tissue items, and they are all currently untreatable and always fatal. Its way of infection is given by a particular alpha and beta protein chain incorrectly folded, which causes other proteins to assume the same abnormal conformation. These proteins are then able to modify adjacent proteins.
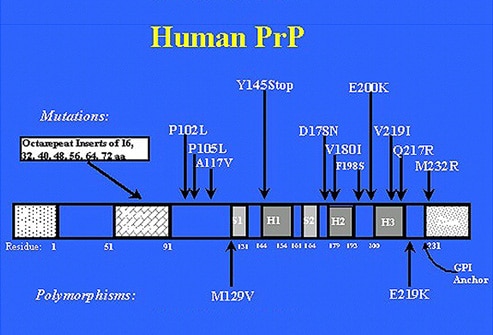
In humans, the PrP is made up of 253 aminoacids, its weigth is about 34 kDa and it is encoded by a single gene that is on the short arm of chromosome 20. The complex of protein maturation involves the reduction of 22 R (residual chain of aminoacids linked) of NH2 and 23 of terminal COOH. Linked to aminoacid 230, there is a molecule of GPI (glycosyl phosphatidyl inositol) which functions to anchor the mature protein to the cell membrane of the neurons.
The prion (PrPres = protease-resistant prion protein or protein PrPsc = scrapie) would be the altered form of a useful protein present in mammals (PrPsen = protein or protease-sensitive cellular protein PrPc), including man, in all organs but in particular on cells of the nervous tissue, whose function hypothesized involves the transmission of nerve impulses and some mechanisms relating to long-term memory.
The prion protein, therefore, exists in two isoforms: PrPc and PrPsc, which have molecular weight and amino acid sequence identical, differ in tertiary and quaternary structure. PrPc (or PrP-sens), already referred to above normal isoform is sensitive to detergents and proteases, and PrPsc (or PrP-res), pathological isoform, partially resistant to detergents and proteases, which when treated with these substances gives rise to the PrP27-30.
PrPsc and PrP27-30 are invariably associated with TSEs (transmissible spongiform encephalopathies) and the isoform PrPC has a polymorphism associated specifically with certain TSEs, including FFI.
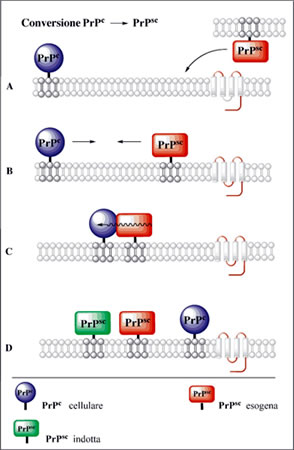
Once anchored to neurons, if it comes into contact with PrPsc, PrPc undergoes a conformational change (the difference between the two is only conformational and is linked to the point mutation of a single amino acid, Met/Val). The PrPsc is slightly soluble and therefore precipitates forming insoluble aggregates called amyloid plaques that trigger the chain reaction that gradually damages the neuronal tissue.
The prion protein (PrPc) is generally harmless. In the benign form, its skeleton folds forming several propellers. PrPc is converted into the infectious form (PrPsc) when most of the skeleton extends, forming the so-called beta strands.
It is demonstrated the existence, within the prion protein, of a structure with remarkable stability when provided at four a-helices, but is also very stable in b-sheet conformation; X-ray diffraction studies have validated the model of the conversion from a-helix to b-sheet too. Furthermore, since the block of protein synthesis leads to a stop of the formation of PrPsc, it is likely that in the transition from PrPc (a-helix) towards PrPsc (b-sheet) is essential to the action of other cellular proteins, most likely chaperonins type.
Some functional studies had shown that a lack of gabaergic inhibition can contribuite to insomnia. In some patients mutations of GABRB3 gene had been found.
NEUROPATHOLOGIC MANIFESTATIONS
In FFI cerebral damages are due to the ammassment of abnormal prions in different encephalic areas.
The neuropathologic manifestations of FFI include thalamic degeneration, with selective involvement of the anterior ventral and mediodorsal thalamic nuclei, inferior olivary and cerebellar changes, and some spongiform change of the cerebral cortex.
The thalamic lesion caused by FFI was already demonstrated in the first cases studied in 1983. Doctor Ignazio Roiter, which cured members of Treviso’s family in which the pathology was identified for the first time, finds out in both brothers a thalamic lesion which leads to its disconnection from hypothalamus. In fact, thalamus,the essential station of reticular ways which active the cerebral cortex to circadian rythm, is usually united with hypothalamus, rilevant for the control of neurovegetative functions and in the promotion of sleep; this is the reason why the disconnection bring to the absence of stop signals to excitatory stimulus directed from thalamus to cortex.
It was suddenly demonstrated that these lesions depended on prion alterations but the relationship between the topography of PrPres (prion protein) and neural dysfunction is still unclear. In FFI, PrPres appears to be more widespread than suggested by lesions or areas of hypometabolic function. Furthermore, the magnitude of PrPres is not correlated with clinical severity, nor does its distribution parallel that of cellular apoptosis. Finally, structures that contain equal amounts of PrPres (such as the thalamus and brainstem) are differentially vulnerable, the thalamus being much more so. All FFI patients show similar amounts of PrPres in the thalamus and brainstem. However, those whose disease runs a short duration show the least amount and most focal distribution of PrPres, with the main accumulation being primarily in limbic areas (eg, entorhinal cortex or the cingulate gyrus) and in subcortical structures (including the thalamus, hypothalamus, and brainstem).With disease of longer duration, the abnormal protein becomes detectable in the cerebral cortex and eventually exceeds the amount observed in subcortical areas.
Thus, two interpretations regarding the role of PrPres are possible. Either PrPres is not toxic and programmed cell death results from the loss of important signals from the normally protective PrNP, or PrPres is toxic but different types of neural tissue are differentially vulnerable to it, in ways related to both tissue type and polymorphic status of C 129.

For more information about the FFI, prions and thalami degeneration
SYMPTOMPS
- Progressive and ferocious insomnia;
- waking sleep;
- hallucinations;
- autonomic disturbances suggestive of sympathetic overdrive;
- a rise in circulating catecholamine levels;
- cognitive changes;
- ataxia;
- endocrine manifestations;
- confusional state.
CLINICAL FEATURES AND HISTORY OF THE DISEASE
It is told in the book of the researcher T.D. Max the story of the first patient affected by Fatal Familiar Insomnia (FFI). In the book "The family that could not sleep" in fact, the author tells the story of an Italian doctor who died in 1765, which would be the first carrier of the genetic mutation that produces the disease (the so-called "patient zero") and reconstructs the path through the generations of man until the 80s.
Max has long studied the family tree of the patient and documents from the eighteenth to twentieth century have allowed the identification of deaths related to IFF generations of the family of the patient zero. Thanks to Silvano, a descendent of the first patient, who died just in the 80s, it was possible to investigate more deeply the causes of fatal familial insomnia. In fact, this man has courageously chosen to donate his body and especially his brain to science so necessary investigations could have been done. Post-mortem analysis of his brain structures made possible to show that the disease process of FFI is a break caused by a genetic mutation of the proteins present in the brain, which, accumulating, end up destroying neurons.
"Within a month, it is easy to see that you have a disease like any other," says Max. "First of all, - he says - you can not sleep anymore. Then, you can not walk, or concentrate on anything. Usually, in the space of nine months, you will die."

Silvano is a 53 year old Venetian extremely energetic and highly educated man who "lived" the reality of dreams and represented them physically. While sleeping he got up, he stopped and gave the military salute. When the family woke him up, told to dream that he was witnessing a coronation. Or he got out of bed, with his arms outstretched toward the floor. If someone asked him where he was, he replied: "At the airport with my bags, I'm leaving."
It might seem normal sleepwalking, except that in that state Silvano would have been 24 hours prisoner until he died nine months after onset, on 21st July 1984.
A few years earlier, two Sandro's sisters exhaled in the same terrible and painful way.
The endocrinologist Ignazio Roiter, explains in an article in the "Corriere della Sera": the difficult research that led him to reconstruct a family tree from 1700 to see if the disease was hereditary, due to the compatibility of the descriptions of the three people's deaths. It was based on the parish archives, testimonies and stories, records of psychiatric hospitals, where most of these patients were interned before the IFF was officially recognized ("Then the asylums were particularly popular, because these patients were taken for fools and then interned" he says).
His was the insight that lack of sleep was not the cause of the disease but its direct effect and the analysis of a portion of the brain of one of Silvano's sisters, with the help of the neuropathologist in Ginevra, Erwin Wildi. But the real watershed in the history of the syndrome, was precisely that Silvano had agreed to be studied by the team of Elio Lugaresi, director of the Sleep Center of Neurology in Bologna. Thanks to the observations on the disease, histological examination of the brain performed in the laboratory of Neuropathology of Cleveland by Pierluigi Gambetti, a Lugaresi's student, and the involvement of the Nobel Prize Stanley Prusiner, it was possible to identify it as a prion disease.
On 16th October 1986, the work of Lugaresi and Gambetti was published in the "New England Journal of Medicine" and fatal familial insomnia officially entered the world of medicine. "Shortly after the alert, neurologists in Bordeaux hospital have been in contact with Bologna. They recognized that perhaps a few years earlier had passed people with these disorders. They literally dug up families, making post-mortem diagnosis of fatal familial insomnia" says Roiter.