DEFINITION
Adenosine deaminase (ADA) deficiency is an inherited disorder that damages the immune system and causes severe combined immunodeficiency (SCID). It is related to an homozygous for a defective gene of Adenosine deaminase (also known as ADA), an enzyme involved in purine metabolism. There are types of SCID:

It is needed for the breakdown of adenosine from food and for the turnover of nucleic acids in tissues.
SCID ADA-deficient.
EPIDEMIOLOGY
Adenosine deaminase deficiency is very rare and is estimated to occur in approximately 1 in 200,000 to 1,000,000 newborns worldwide. This disorder is responsible for approximately 15 percent of SCID cases.
SYMPTOMS
If a baby exhibits any of the following persistent symptoms within the first year of life, he or she should be evaluated for SCID or other types of immune deficiency syndromes:
-Eight or more ear infections
-Two or more cases of pneumonia
-Infections that do not resolve with antibiotic treatment for two or more months
-Failure to gain weight or grow normally
-Infections that require intravenous antibiotic treatment
-Deep-seated infections, such as pneumonia that affects an entire lung or an abscess in the liver
-Persistent thrush in the mouth or throat
-A family history of immune deficiency or infant deaths due to infections
:http://www.annualreviews.org/doi/abs/10.1146/annurev.immunol.22.012703.104614
DIAGNOSIS
-blood exams (t-cells counts)
-x-ray (absence of thymic shadow, lung abnormalities, skeletal abnormalities)
PATHOGENESIS
The ADA gene provides instructions for producing the enzyme adenosine deaminase. This enzyme is found throughout the body but is most active in lymphocytes, the specialized white blood cells that protect the body against potentially harmful invaders by making antibodies or by directly attacking virus-infected cells. Lymphocytes are produced in specialized lymphoid tissues including the thymus, which is a gland located behind the breastbone, and lymph nodes, which are found throughout the body. Together, lymphocytes in the blood and in lymphoid tissues make up the immune system.
The function of the adenosine deaminase enzyme is to eliminate a molecule called deoxyadenosine, which is generated when DNA is broken down. Adenosine deaminase converts deoxyadenosine, which is toxic to lymphocytes, to another molecule called deoxynosine that is not harmful. Mutations in the ADA gene reduce or eliminate the activity of adenosine deaminase and allow the buildup of deoxyadenosine to levels that are toxic to lymphocytes.
Immature lymphocytes in the thymus are particularly vulnerable to a toxic buildup of deoxyadenosine. The number of lymphocytes in other lymphoid tissues is also greatly reduced, resulting in the signs and symptoms of SCID.
Children with SCID lack virtually all immune protection from bacteria, viruses, and fungi. They are prone to repeated and persistent infections that can be very serious or life-threatening. These infections are often caused by "opportunistic" organisms that ordinarily do not cause illness in people with a normal immune system. Infants with SCID typically experience pneumonia, chronic diarrhea, and widespread skin rashes, and grow much more slowly than healthy children. Some also have neurological problems such as developmental delay, movement disorders, and hearing loss. If not treated in a way that restores immune function, children with SCID usually live only a year or two.
Most individuals with adenosine deaminase deficiency are diagnosed with SCID in the first six months of life. In about 10 percent to 15 percent of cases, onset of immune deficiency is delayed to between 6 and 24 months of age (delayed onset) or even until adulthood (late onset). Immune deficiency in these later-onset cases tends to be less severe, causing primarily recurrent upper respiratory and ear infections. Over time, affected individuals may develop chronic lung damage, allergies, and other health problems.
The enzyme adenosine deaminase is encoded by a gene on chromosome 20. ADA deficiency is inherited in an autosomal recessive manner. This means the defective gene responsible for the disorder is located on an autosome (chromosome 20 is an autosome), and two copies of the defective gene (one inherited from each parent) are required in order to be born with the disorder. The parents of an individual with an autosomal recessive disorder both carry one copy of the defective gene, but usually do not experience any signs or symptoms of the disorder.
the role of adenosine in causing the immunodeficiency associated with adenosine deaminase deficiency
PATIENT RISK FACTORS
Vascular
Genetic
Acquired
Hormonal
Genetic
Acquired
TISSUE SPECIFIC RISK FACTORS
anatomical (due its structure)
vascular (due to the local circulation)
physiopathological (due to tissue function and activity)
COMPLICATIONS
THERAPY
The goal of all forms of ADA-SCID therapy is to return the immune system to functioning order.
-Enzyme replacement therapy (ERT) has been used successfully in the treatment of SCID for more than 20 years. ADA is obtained from cows, purified, and pegylated to produce Adagen. Pegylation attaches the molecule polyethylene glycol (PEG) to the ADA enzyme, which helps it to remain in circulation in the body, by disguising it from the immune system and avoiding antibody formation. ADAGEN® (pegademase bovine) Injection is indicated for adenosine deaminase (ADA) deficiency in patients with severe combined immunodeficiency disease (SCID) who are not suitable candidates for – or who have failed – bone marrow transplantation.
 How i treat Ada deficiency
How i treat Ada deficiency
-Bone Marrow Transplant (BMT) is also known as hematopoietic stem cell transplantation, or HSCT. When the cells are from another person and not the patient’s identical twin, it is referred to as an allogeneic HSCT. Donor bone marrow cells are given to the patient in order to provide them with normal hematopoietic stem cells (HSCs). HSCs exist in the bone marrow and produce new blood cells, including the T lymphocytes. A successful BMT can restore T-lymphocyte function, providing a cure for patients with ADA-SCID. Potential stem cell donors for ADA-SCID patients would ideally be an HLA-matched sibling or other family member, or else a matched unrelated donor. The success rate for BMT with a matched donor ranges as high as 90%. For those without a matched sibling, it may be possible to find a matched, unrelated donor through medical databases.
In recent years, BMT techniques have been developed that also offer a chance of success with a mismatched donor. However, mismatched donors usually offer lower rates of success. One reason for this is that the patient must be treated with chemotherapy before receiving the mismatched stem cells. The choice of chemotherapy regimen may vary depending on the patient and the treatment protocols of the treatment center.
":http://arapaho.nsuok.edu/~castillo/NotesImages/Topic140NotesImage1.jpg
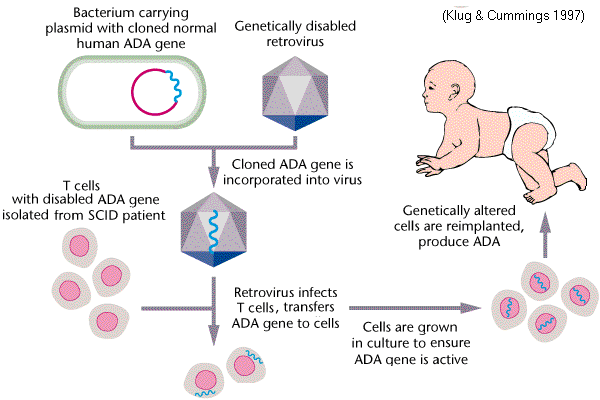
-Gene therapy is currently an experimental treatment for ADA-SCID. Since ADA-SCID is an inherited disorder caused by a defect in the genetic code of the patient, the goal of gene therapy is to correct the defective genes so that the immune system can be restored.
To implement gene therapy, a sample of cells from the patient’s defective bone marrow is taken to a medical laboratory. In the lab, a functional ADA gene is inserted into a specially engineered retroviral vector, and then introduced into the cell. Once the normal ADA gene has integrated with the bone marrow cells, the cells are injected back into the patient’s bone marrow, where they may engraft, proliferate, and gradually replace the defective cells. Research to develop the ideal treatment regimen continues. In some clinical studies, patients were treated with chemotherapy before receiving the gene therapy. The researchers believe that this extra step prevented the patients’ bodies from rejecting the new cell graft. The most recent medical data indicate that patients may have a better chance of success with gene therapy when they discontinue taking PEG-ADA prior to receiving the GT. This may be because the absence of PEG-ADA provides the genetically modified cells with a survival advantage.
CASE STUDY
Between the years of 1968 and 1973 doctors were occasionally able to diagnosis a case of SCID, however, if there was no HLA matched sibling they were unable to correct the defect. Unfortunately, most of these cases were not diagnosed until the child was critically ill. In 1971 an unusual opportunity occurred. A mother who had lost a child to SCID was pregnant with another boy. Since the risk of another affected child was a real possibility, plans were made on how to keep this child healthy until his immune system could be corrected. David was born on September 21, 1971 and was immediately placed into a specially designed isolator crib where the air was specially filtered and all items which went into the crib were sterilized. It was quickly proven that his immune system was indeed defective, but there was hope that his older sister would be a match for a bone marrow transplant. Unfortunately, his sister was not a good match and at that time there were no donor registries. The rest of the family was tested, but no one was a match for David. Consequently, as he grew, so did his bubbles. The media during that time chronicled his life and he became known to the world as the "Bubble Boy".
It wasn't until the early 80's that a technique was developed to use a bone marrow donor who was less than a 100% match. This method eventually made it possible to use a haploidentical or 1/2 matched donor for a successful BMT, which has been and remains the most significant treatment development for this condition over the past two decades. David died in 1984 following an unsuccessful bone marrow transplant, an attempt to provide him with the capacity to fight infections on his own and thus free him from the bubble.
Gene therapy: treating the bubble
Margherita Saettone (735727)