Bile Introduction
Bile is a complex fluid containing water, electrolytes and a battery of organic molecules including bile acids, cholesterol, phospholipids and bilirubin that flows through the biliary tract into the small intestine. There are two fundamentally important functions of bile in all species:
• Bile contains bile acids, which are critical for digestion and absorption of fats and fat-soluble vitamins in the small intestine.
• Many waste products, including bilirubin, are eliminated from the body by secretion into bile and elimination in feces.
Adult humans produce 400 to 800 ml of bile daily, and other animals proportionately similar amounts. The secretion of bile can be considered to occur in two stages:
• Initially, hepatocytes secrete bile into canaliculi, from which it flows into bile ducts. This hepatic bile contains large quantities of bile acids, cholesterol and other organic molecules.
• As bile flows through the bile ducts it is modified by addition of a watery, bicarbonate-rich secretion from ductal epithelial cells.
In species with a gallbladder (man and most domestic animals except horses and rats), further modification of bile occurs in that organ. The gall bladder stores and concentrates bile during the fasting state. Typically, bile is concentrated five-fold in the gall bladder by absorption of water and small electrolytes - virtually all of the organic molecules are retained.
Secretion into bile is a major route for eliminating cholesterol. Free cholesterol is virtually insoluble in aqueous solutions, but in bile, it is made soluble by bile acids and lipids like lethicin. Gallstones, most of which are composed predominantly of cholesterol, result from processes that allow cholesterol to precipitate from solution in bile.
Role of Bile Acids in Fat Digestion and Absorption
Bile acids are derivatives of cholesterol synthesized in the hepatocyte. Cholesterol, ingested as part of the diet or derived from hepatic synthesis is converted into the bile acids cholic and chenodeoxycholic acids, which are then conjugated to an amino acid (glycine or taurine) to yield the conjugated form that is actively secreted into cannaliculi.
Bile acids are facial amphipathic, that is, they contain both hydrophobic (lipid soluble) and polar (hydrophilic) faces. The cholesterol-derived portion of a bile acid has one face that is hydrophobic (that with methyl groups) and one that is hydrophilic (that with the hydroxyl groups); the amino acid conjugate is polar and hydrophilic.
Their amphipathic nature enables bile acids to carry out two important functions:
• Emulsification of lipid aggregates: Bile acids have detergent action on particles of dietary fat which causes fat globules to break down or be emulsified into minute, microscopic droplets. Emulsification is not digestion per se, but is of importance because it greatly increases the surface area of fat, making it available for digestion by lipases, which cannot access the inside of lipid droplets.
• Solubilization and transport of lipids in an aqueous environment: Bile acids are lipid carriers and are able to solubilize many lipids by forming micelles - aggregates of lipids such as fatty acids, cholesterol and monoglycerides - that remain suspended in water. Bile acids are also critical for transport and absorption of the fat-soluble vitamins.
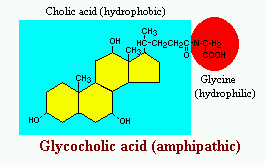
Figure: R.Bowen
Role of Bile Acids in Cholesterol Homeostasis
Hepatic synthesis of bile acids accounts for the majority of cholesterol breakdown in the body. In humans, roughly 500 mg of cholesterol are converted to bile acids and eliminated in bile every day. This route for elimination of excess cholesterol is probably important in all animals, but particularly in situations of massive cholesterol ingestion.
Interestingly, it has recently been demonstrated that bile acids participate in cholesterol metabolism by functioning as hormones that alter the transcription of the rate-limiting enzyme in cholesterol biosynthesis.
Enterohepatic Recirculation
Large amounts of bile acids are secreted into the intestine every day, but only relatively small quantities are lost from the body. This is because approximately 95% of the bile acids delivered to the duodenum are absorbed back into blood within the ileum.
Venous blood from the ileum goes straight into the portal vein, and hence through the sinusoids of the liver. Hepatocytes extract bile acids very efficiently from sinusoidal blood, and little escapes the healthy liver into systemic circulation. Bile acids are then transported across the hepatocytes to be resecreted into canaliculi. The net effect of this enterohepatic recirculation is that each bile salt molecule is reused about 20 times, often two or three times during a single digestive phase.
It should be noted that liver disease can dramatically alter this pattern of recirculation - for instance, sick hepatocytes have decreased ability to extract bile acids from portal blood and damage to the canalicular system can result in escape of bile acids into the systemic circulation. Assay of systemic levels of bile acids is used clinically as a sensitive indicator of hepatic disease.
Pattern and Control of Bile Secretion
The flow of bile is lowest during fasting, and a majority of that is diverted into the gallbladder for concentration. When chyme from an ingested meal enters the small intestine, acid and partially digested fats and proteins stimulate secretion of cholecystokinin and secretin. As discussed previously, these enteric hormones have important effects on pancreatic exocrine secretion. They are both also important for secretion and flow of bile:
• Cholecystokinin: The name of this hormone describes its effect on the biliary system - cholecysto = gallbladder and kinin = movement. The most potent stimulus for release of cholecystokinin is the presence of fat in the duodenum. Once released, it stimulates contractions of the gallbladder and common bile duct, resulting in delivery of bile into the gut.
• Secretin: This hormone is secreted in response to acid in the duodenum. Its effect on the biliary system is very similar to what was seen in the pancreas - it simulates biliary duct cells to secrete bicarbonate and water, which expands the volume of bile and increases its flow out into the intestine.
Mechanisms of Bile Formation

Figure:JL Boyer
The formation of bile is a unique and vital function of the liver. Failure to form bile results in progressive cholestatic liver injury and death. Bile is a complex aqueous secretion composed of ~95% water and endogenous solid constituents consisting of bile salts, phospholipid and cholesterol, amino acids, steroids, enzymes, porphyrins, vitamins, and heavy metals as well as exogenous drugs, xenobiotics and toxins. Bile salts are the major organic solute in bile and are necessary for the emulsification and digestive absorption of dietary lipids. Bile serves to eliminate potentially harmful organic lipophilic substances, including xenobiotics and toxins as well as endogenous substrates such as bilirubin and bile salts, not easily excreted by the kidney. Bile is also the major route of excretion for cholesterol. Disorders that impair the production of bile result in the syndrome known as cholestasis. The primary secretion of bile is generated by osmotic gradients that are formed within the bile canaliculus of the hepatocytes by energy dependent mechanisms. The creation of these osmotic gradients depends upon the function of a number of transporting polypeptides that are located on basolateral and apical plasma membrane domains. This primary secretion of bile is subsequently modified by the bile duct epithelium where transport systems in the luminal membranes of cholangiocytes both secrete and absorb certain biliary constituents.
Signal Transduction in Bile Formation and Cholestasis

Figure: M. Sawkat Anwer and Cynthia R.L. Webster
Bile formation involves vectorial transport of solutes from blood to bile and is dependent on coordinated activities of various solute transporters located at the basolateral and apical membranes of hepatocytes and cholangiocytes. Cholestasis results when the vectorial transport of solutes destined for bile is compromised. Our understanding of various transporters, their substrates and locations has increased steadily as is the cellular mechanism regulating these transporters. It is becoming more evident that choleretic and cholestatic agents modify the function of these transporters through various signal tranduction pathways. Cyclic AMP, acting via PKA and the PI3K signaling pathway, stimulate transhepatic transport of bile acids by translocating Ntcp and Bsep to the sinusoidal and the canalicular membrane, respectively. Cell swelling induced by hypo-osmotic media stimulates hepatic uptake and biliary excretion of bile acids via the PI3K and the ERK signaling pathway, respectively. TUDC and cell swelling also stimulate biliary bile acid excretion via the p38 MAPK signaling pathway. TUDC reverses TLC cholestasis by stimulating PKC-mediated translocation of Mrp2 to the canalicular membrane. Calcium, acting via Ca2+/calmodulin-dependent kinases/phosphatases, augments cAMP-mediated translocation of Ntcp, increases tight-junctional permeability by phosphorylating myosin light-chain and stimulates sinusoidal Na+/H+ exchange. PKC stimulates bile acid secretion, most likely by phosphorylating Bsep, and Na+/H+ exchange. However, our understanding of the role of specific PKC isoforms involved in bile formation is still lacking. Molecular mechanisms by which PI3K and MAPK signaling pathways stimulate transporter translocation along the cytoskeleton have not been elucidated. Our knowledge of the role of protein phosphatases in bile formation is rather limited.
Hepatocellular Transport Systems: Basolateral Membrane

Figure: B Hagenbuch and PJ Meier
The basolateral membrane of hepatocytes is equipped with efficient transport systems for uptake of bile salts (Ntcp/NTCP) and xenobiotics (Oatps/OATPs). Under physiological conditions these transporters are important for ongoing bile formation and for efficient hepatic detoxification. Especially Oatps/OATPs seem to play an important role in the hepatic first-pass clearance of xenobiotics and can influence the bioavailability of certain drugs. Under cholestatic conditions these uptake systems are down-regulated to prevent an overflow of hepatocytes with potentially toxic compounds. Furthermore an efficient efflux system is expressed (Mrp3/MRP3) at the basolateral membrane that helps to protect liver cells from intracellular accummulation of toxic bile salts and biotransformation products. Hence, the correct interplay of regulatory mechanisms that control the expression of basolateral transporters is vital for the adaptation of efficient hepatocellular functions to various physiological and pathophysiological conditions.
The ABC of Canalicular Transport
Bile formation is a regulated process and depends on the coordinated action of a number of transporter proteins in the sinusoidal (basal) and canalicular (apical) domains of the hepatocyte. The secretion of substances in the canalicular lumen is mediated by a set of ATP-dependent transport proteins. Dysfunction of any of these proteins leads to retention of substrates, with e.g., conjugated hyperbilirubinemia or cholestasis as a result. In recent years many of the transport proteins involved in bile formation have been identified, cloned, and functionally characterized. Bile salts are highly conserved by means of enterohepatic circulation. At the basolateral membrane the sodium taurocholate cotransporter (NTCP/SLC10A1)
represents the primary carrier for conjugated bile salts such as taurocholate. The OATP/SLC21A family of transporters constitute a family of sodium independent multispecific transporters involved in the uptake of bile salts, organic anions and other amphipathic organic solutes. Following extraction from the portal blood, bile salts are shuttled to the canalicular membrane. At the canalicular membrane, bile salt secretion represents the major and rate-limiting driving force of bile flow. This ATP-dependent secretory process is mediated primarily by the bile salt export pump (BSEP/ABCB11) although studies with Bsep / mice direct towards the existence of a second canalicular bile salt transport system. A major determinant of the bile salt independent bile flow, reduced glutathione, is excreted into the bile canaliculus in an ATP-dependent fashion by the multidrug resistance associated protein 2 (MRP2/ABCC2) which also secretes a variety of organic anions such as leukotrienes, conjugated bilirubin and glutathione conjugates. A chloride/bicarbonate anion exchanger 2 (AE2) is present in the canalicular membrane and facilitates bicarbonate excretion thereby contributing as a second component of the bile salt independentbile flow. Additional MRP isoforms such as MRP1/ABCC1 and MRP3/ABCC3 are present in low abundance in the basolateral membrane. Once secreted into the canalicular space, bile salts appear to partition into the outer leaflet of the canalicular membrane to facilitate the extraction of lipids from the canalicular membrane to form vesicles. Replenishment of the canalicular membrane with phosphatidylcholines appears to be mediated by a specific phosphatidylcholine transfer protein (PCTP) followed by an ATP-dependent phosphatidylcholine translocation from the inner to the outer canalicular membrane. This has been shown to critically depend on the expression of MDR3/ABCB4. Biliary cholesterol secretion may also be facilitated by an ATP-dependent transport mechanism, ABCA1 and ABCG5/ABCG8 representing candidate transport proteins expressed in the canalicular membrane. Plasma lipoproteins such as HDL represent a major source of biliary cholesterol. Following basolateral uptake of HDL-cholesterol via the HDL receptor SRBI, cholesterol trafficking to the canalicular plasma membrane appears to be facilitated by cytosolic sterol carrier protein 2 (SCP2) and liver fatty acid binding protein (FABP1). The composition of hepatic bile is modulated by transport proteins present in cholangiocytes. The cystic fibrosis transmembrane regulator (CFTR/ABCC7) facilitates chloride entry into bile whereas the anion exchanger 2 (AE2) facilitates bicarbonate secretion. Two other membrane proteins of cholangiocytes are involved in bile formation. This includes i) the apical bile salt transporter IBAT/SLC10A2 which is also expressed in the ileum and appears to be involved in cholehepatic shunting of bile salts and ii) FIC1/ATP8B1, a P-type ATPase which represents a putative aminophospholipid transporter in the apical membrane of cholangiocytes and hepatocytes.
Transcription regulation of hepatobiliry transporters
The expression and activities of hepatobiliary transporter genes is a critical component of liver function. Both sinusoidal and canalicular membrane transporters are responsible for the coordinated transport of a wide variety of organic anions, drugs, toxins, endobiotics and bile acids that ultimately are metabolized by hepatocytes and secreted into bile. Over the past decade, a tremendous amount of new information has been uncovered regarding the molecular identification of hepatobiliary transporter genes responsible for the delivery of the principal solutes in bile, and therefore the mechanisms underlying the generation of bile. Along with the cloning and identification of critical hepatobiliary transporter genes has come the capability of exploring, and possibly modifying, the molecular mechanisms driving alterations in transporter gene expression in health and disease. Regulation at the level of mRNA transcription initiation is becoming increasingly recognized as the predominant means governing transporter gene expression. An expanding role for Hepatocyte Nuclear Factor 1a and several members of the nuclear receptor superfamily of ligand-regulatable transcription factors permits coordination of expression of multiple transporter genes. In particular, the recently-identified nuclear receptor for bile acids, FXR, serves as both sensor and effector to help maintain intracellular bile acid homeostasis. These findings have significant importance in our current understanding, and as a means of directing future therapy, of liver diseases where cholestasis is a prominent feature.
Cholestasis: An Intracellular "Traffic Jam"
Mutations in the coding region of BSEP which result in its absence from the bile canalicular membrane are manifested by progressive cholestasis and liver damage. Defects in intracellular trafficking and/or posttranslational regulation of BSEP may produce a similar phenotype. To test this hypothesis, it was necessary to determine the pathway utilized by BSEP and other canalicular ABC transporters and its regulation. These studies reveal that BSEP traffics from Golgi to a subapical recycling endosomal compartment from which the transporter cycles to and from the canalicular membrane; the efflux compartment is cAMP-dependent. A second intracellular pool which responds to taurocholate was identified. Both intracellular sites contain at least six times more BSEP as is present in the canalicular membrane. Using pulse-chase and subcellular fractionation studies in rats and on-line image analysis of the movement of chimeric fluorescent BSEP in WIFb cells, the trafficking rates were measured and the critical role of 3´ phosphoinositide products of phosphatidyl (PI) 3-kinase, microtubules and other intracellular components was demonstrated. In addition, the activity of BSEP in the canalicular membrane is regulated by PI 3-kinase products and by cAMP. All of these effects occur in the absence of new protein synthesis. Studies indicate that the hepatocyte is protected from the detergent effects of retained bile acids by post-translationally mediated enhanced BSEP transcellular transport and activation within the canalicular membrane. Experimentally induced alterations in trafficking components impaired BSEP delivery to and/or activation in the canalicular membrane. The results suggest that intrahepatic cholestasis may result from an intracellular traffic jam and create exciting opportunities to investigate the interaction between inheritable defects in ABC transporters and acquired conditions in the pathogenesis of cholestasis associated with drugs, viruses, metals and other factors.
The biology of cholangiocytes
Cholangiocytes are the epithelial cells of the bile duct. They are cuboidal epithelium in the small interlobular bile ducts, but become columnar and mucus secreting in larger bile ducts approaching the porta hepatis and the extrahepatic ducts.
In the healthy liver, cholangiocytes contribute to bile secretion via net release of bicarbonate and water. Several hormones and locally acting mediators are known to contribute to cholangiocyte fluid/electrolyte secretion. These include secretin, acetylcholine, ATP, and bombesin.
Cholangiocytes act through bile-acid independent bile flow, which is driven by the active transport of electrolytes. In contrast, hepatocytes secrete bile through bile-acid dependent bile flow, which is coupled to canalicular secretion of bile acids via ATP-driven transporters. This results in passive transcellular and paracellular secretion of fluid and electrolytes through an osmotic effect.
Importantly, cholangiocytes are the target of disease in a variety of conditions often known as "cholangiopathies". These diseases include primary biliary cirrhosis, primary sclerosing cholangitis, AIDS cholangiopathy, disappearing bile duct syndromes, Alagille's syndrome, cystic fibrosis, and biliary atresia. As a group, cholangiopathies account for approximately 18% of adult liver transplantations and the majority of pediatric liver transplantations.
Active scientific investigation of cholangiocytes focuses on such diverse processes as mechanisms of fluid/electrolyte secretion, regulation of cholangiocyte proliferation, roles of cholangiocytes in the pathogenesis of liver fibrosis and cirrhosis, and cholangiocyte apoptosis. Specific investigation of individual cholangiopathies is also pursued actively.
Fat Absorption and Lipid Metabolism in Cholestasis
The liver has a central role in control of various aspects of lipid metabolism. Primarily, the liver produces bile, constituents of which are required for efficient intestinal fat absorption. Additionally, biliary secretion of cholesterol (either as such, or after metabolism in the form of bile salts) and phospholipids from the liver into the intestine is of major importance in body lipid homeostasis. The liver is the major source of plasma lipoproteins: it synthesizes apoproteins (i.e., apo A-I, apo B, apo E) that regulate many complex metabolic interconversions between lipoprotein classes, as well as lipoprotein lipid constituents as cholesterol, triglycerides and phospholipids. The liver is also the major site of clearance of circulating lipoproteins, which are subsequently catabolized in the hepatocytes. Additionally, the liver synthesizes enzymes (e.g., LCAT, CETP, PLTP, LPL) which are involved in lipoprotein metabolism in the plasma compartment. Finally, the liver is the site of active synthesis, metabolism and/or oxidation of various lipid classes, including long-chain polyunsaturated fatty acids. In view of this multitude of essential functions that are in part strongly interrelated, it is evident that disturbances in bile formation in cholestatic liver disease will have a strong impact on various aspects of lipid metabolism in the body. Consequences of cholestasis, which is functionally defined as decreased or absent bile flow from the liver into the intestine, may be related to: 1. the absence of specific bile components at their sites of action, particularly in the intestine 2. disruption of the continuous flux of lipids from the liver into bile and intestine, resulting in accumulation of toxic and non-toxic bile components in the body, most notably in hepatocytes, with concomitant alterations in hepatocyte function. 3. characteristic alterations in plasma lipoprotein composition associated with cholestatic liver diseases such as decreased HDL levels and the appearance of lipoprotein X.
Acquired Alterations in Transporter Expression and Function in Cholestasis
Exposure to cholestatic injury (e.g., drugs, hormones, proinflammatory cytokines, biliary obstruction/destruction), hereditary mutations of transporter genes, or the combination of both result in decreased expression and function of hepatobiliary transport systems. These molecular changes may contribute to impaired hepatic uptake and excretion of bile salts and other organic anions (e.g., bilirubin) in cholestasis. In addition, alterations in transporter expression may represent secondary and adaptive changes, limiting the accumulation of potentially toxic biliary constituents in the cholestatic liver by providing alternative excretory routes. The mediators and molecular mechanisms responsible for changes in transporter expression are being increasingly understood at transcriptional and post-transcriptional levels. The molecular changes of hepatobiliary transport systems in cholestasis may represent a potential target for specific therapeutic interventions aimed at restoration of defective hepatobilary transporter expression and stimulation of adaptive rescue pathways.
Pathology of Cholestasis
Cholestasis formation may give rise to extensive parenchymal changes, without significant alterations in biliary tree morphology. Sepsis or systemic inflammatory conditions also can cause severe hepatocellular cholestasis without obstruction, although there may be attendant inflammatory changes in portal tracts. There is a striking set of bile duct diseases leading eventually to destruction of the intrahepatic and/or extrahepatic biliary tree, with characteristic and oft-times diagnostic histologic features evident on percutaneous liver biopsy. While non-obstructive cholestasis may be relatively mild and reversible depending upon the underlying cause, obstructive cholestasis will lead to substantial hepatic compromise from retained bile. Because of the central role of bile formation in normal hepatic function, and the severe hepatic and systemic toxicity of relentless bile secretory failure, there is a great need to correct the cause of cholestasis if at all possible. Morphological assessment of liver tissue, either through percutaneous liver biopsy or open exploration with biopsy, can play a major role in the clinical assessment and monitoring of these patients. The morphology of cholestasis is also a major clue to the underlying etiologies of disease.
Bile Acid-Mediated Apoptosis in Cholestasis
Toxic bile acids induce apoptosis by activating cell surface membrane death receptors. The activated death receptors stimulate a signaling cascade involving the pro-apoptotic Bcl-2 poteins Bid and Bax. The proapoptotic Bcl-2 proteins initiate mitochondrial injury, the mitochondrial permeability transition (MPT) and oxidative stress. Inhibition of death receptors and their cascades may prove useful in attenuating liver injury during cholestasis.
Disorders of Bile Acid Transport
Bile salts take part in a rather efficient enterohepatic circulation in which most of the secreted bile salts are reclaimed by absorption in the terminal ileum. In the liver the sodium dependent taurocholate transporter (NTCP) at the basolateral (sinusoidal) membrane and the bile salt export pump (BSEP) at the canalicular membrane mediate hepatic uptake and hepatobiliary secretion of bile salts. Transporter genes are transcriptionally regulated by members of the nuclear hormone receptor family. These are ligand activated transcription factors that respond to bile salts. At high bile salt concentrations the expression of NTCP is reduced and that of BSEP increased. Canalicular secretion is the dominant element in the enterohepatic cycling of bile salts and most genetic diseases are caused by defects of canalicular secretion. Progressive familial intrahepatic cholestasis results from mutations in the FIC1 gene. This, in a not well understood way, leads to relasping or permanent cholestasis. The relapsing disease is called benign recurrent intrahepatic cholestasis (BRIC), the permanent cholestasis PFIC type 1. Byler disease is a PFIC type 1 disease. PFIC type 2 results from mutations in the BSEP gene. This leads to permanent cholestasis since birth.PFIC type 1 and type 2 are characterized by a low to normal serum gamma-glutamyltransferase activity. Bile diversion procedures, causing a decreased bile salt pool, have a beneficial effect in a number of patients with these diseases. However, liver transplantation is often necessary. PFIC type 3 is caused by mutations in the MDR3 gene. MDR3 is a phospholipid translocator in the canalicular membrane. Because of the inability to secrete phospholipids, patients with PFIC type 3 produce bile acid-rich toxic bile that damages the intrahepatic bile ducts. Ursodeoxycholic acid therapy is useful for patients with a partial defect. Liver transplantation is a more definite therapy for these patients. Among patients with intrahepatic cholestasis of pregnancy, heterozygosity for MDR3 gene mutations is frequently found. In patients with MDR3 defects, the serum gamma-glutamyltransferase activity is elevated.
Genetic Defects in Biliary Lipid Transport
In terms of solute mass, lipids are the second most important component of bile. Biliary lipids mainly consist of phospholipid (almost exclusively phosphatidylcholine; PC) and cholesterol. The ratio in which these two lipids are secreted varies considerably between species. In rodents the phospholipid over cholesterol ratio is 5-10 while in man this is considerably lower. The relatively high concentration of cholesterol in human bile represents the major risk factor to gallstone formation. In the past years it has become clear that biliary lipid secretion serves an important function in defending hepatocytes and bile duct epithelial cells against bile salt induced toxicity. The secretion of biliary lipids is a complex process that we only poorly understand. It is clear, however, that it involves translocation of phosphatidylcholine across the canalicular membrane by MDR3 P-glycoprotein (Pgp) and that the actual secretion process is driven by bile salt present in the canalicular lumen. Patients lacking MDR3 P-glycoprotein develop progessive liver disease. It is becoming clear that other lipid translocation processes occur in the canalicular membrane which may play a role in biliary lipid and bile salt secretion.
Transport of Bilirubin and its Conjugates across Hepatocellular Membrane Domains and the Conjugated Hyperbilirubinemia of Dubin-Johnson Syndrome
Bilirubin, the end product of heme catabolism, needs to be taken up into hepatocytes and is then glucuronidated within the cells prior to its excretion via bile. Members of the SLC21A family in the sinusoidal membrane of hepatocytes selectively mediate the uptake of unconjugated bilirubin and of bilirubin conjugates. After conjugation within the hepatocyte by UGT1A1, yielding monoglucuronosyl and bisglucuronosyl bilirubin, the conjugates are transported across the canalicular membrane into bile by the apical conjugate export pump MRP2, a member of the ABCC subfamily of ATP-dependent transporters. MRP2 also mediates the export of a number of other amphiphilic anions and anionic substances including xenobiotics conjugated with glutathione, glucuronate, or sulfate. Mutations in the ABCC2 gene leading to the absence of a functional MRP2 protein from the canalicular membrane, are the molecular basis of Dubin-Johnson syndrome in humans. Two hyperbilirubinemic rat strains with a hereditary Mrp2 deficiency may be considered as animal models for Dubin-Johnson syndrome and have been helpful in the molecular identification and functional characterization of MRP2. A number of single nucleotide polymorphisms has been recently described in the ABCC2 gene, however, the functional consequences, if any, of each of the polymorphism still await clarification. A transitory MRP2 deficiency with MRP2 being retrieved into subapical vesicles is observed under cholestatic conditions in rats and humans. Under conditions of hereditary or acquired MRP2 deficiency, the isoform MRP3 is upregulated in the sinusoidal membrane of hepatocytes. This mechanism may be more pronounced in rat than in human liver. MRP3 mediates the export of bilirubin conjugates and other anionic conjugates with glucuronate or sulfate into blood. Recent results show that the expression of MRP2 is regulated by several nuclear receptor-mediated pathways including the FXR, the PXR, and the CAR receptor.
Cholestasis
Cholestasis is a condition caused by rapidly developing (acute) or long-term (chronic) interruption in the excretion of bile (a digestive fluid that helps the body process fat). The term is taken from the Greek chole, bile, and stasis, standing still.
Cholestasis is caused by obstruction within the liver (intrahepatic) or outside the liver (extrahepatic). The obstruction causes bile salts, the bile pigment bilirubin, and fats (lipids) to accumulate in the blood stream instead of being eliminated normally.
Intrahepatic cholestasis is characterized by widespread blockage of small ducts or by disorders, such as hepatitis, that impair the body's ability to eliminate bile. Extrahepatic cholestasis can occur as a side effect of many medications. It can also occur as a complication of surgery, serious injury, tissue-destroying infection, or intravenous feeding. Extrahepatic cholestasis can be caused by conditions such as tumors and gallstones that block the flow of bile from the gallbladder to the first part of the small intestine (duodenum).
Pregnancy increases the sensitivity of the bile ducts to estrogen, and cholestasis often develops during the second and third trimesters of pregnancy. This condition is the second most common cause of jaundice during pregnancy, but generalized itching (pruritus gravidarum) is the only symptom most women experience. Cholestasis of pregnancy tends to run in families. Symptoms usually disappear within two to four weeks after the baby's birth but may reappear if the woman becomes pregnant again.
A similar condition affects some women who take birth control pills. Symptoms disappear after the woman stops using oral contraceptives. This condition does not lead to chronic liver disease. A woman who develops cholestasis from either of these causes (pregnancy or birth control hormones) has an increased risk of developing cholestasis from the other.
Benign familial recurrent cholestasis is a rare condition characterized by brief, repeated episodes of itching and jaundice. Symptoms often disappear. This condition does not cause cirrhosis.
Drug-induced cholestasis may be a complication of chemotherapy or other medications. The two major types of drug-induced cholestasis are direct toxic injury and reactions unique to an individual (idiosyncratic reactions). In direct toxic injury, the severity of symptoms parallels the amount of medication involved. This condition:
* develops a short time after treatment begins
* follows a predictable pattern
* usually causes liver damage
Direct toxic reactions develop in 1% of all patients who take chlorpromazine (Thorazine), a tranquilizer and antinausea drug. Idiosyncratic reactions may occur at the onset of treatment or at a later time. Allergic responses are varied and are not related to the amount of medication being taken.
Causes and symptoms
Intrahepatic cholestasis is usually caused by hepatitis or by medications that can produce symptoms resembling hepatitis. Phenothiazine-derivative drugs, including chlorpromazine, can cause sudden fever and inflammation. Symptoms usually disappear after use of the drug(s) is stopped. In rare cases, a condition resembling chronic biliary cirrhosis (a progressive disease characterized by destruction of small bile ducts) persists even after the medication is stopped. Some patients experience a similar reaction in response to tricyclic antidepressants (amitriptyline, imipramine), phenylbutazone (Butazolidin), erythromycin estolate (Estomycin, Purmycin), and other drugs. Intrahepatic cholestasis may also be caused by alcoholic liver disease, primary biliary cirrhosis, cancer that has spread (metastasized) from another part of the body, and a number of rare disorders.
Extrahepatic cholestasis is most often caused by a stone obstructing the passage through which bile travels from the gallbladder to the small intestine (common bile duct) or by pancreatic cancer. Less often, the condition occurs as a result of non-cancerous narrowing of the common duct (strictures), ductal carcinoma, or disorders of the pancreas.
Cholestasis caused by the use of steroids causes little, if any, inflammation. Symptoms develop gradually and usually disappear after the drug is discontinued; but also other drugs can cause cholestasis.
Symptoms of both intrahepatic and extrahepatic cholestasis include a yellow discoloration of the skin (jaundice), dark urine, and pale stools. Itching over the skin may be severe if the condition is advanced.
Symptoms of chronic cholestasis include:
* skin discoloration
* scars or skin injuries caused by scratching
* bone pain
* yellowish fat deposits beneath the surface of the skin (xanthoma) or around the eyes (xanthelasma)
Patients with advanced cholestasis feel ill, tire easily, and are often nauseated. Abdominal pain and such systemic symptoms as anorexia, vomiting, and fever are usually due to the underlying condition that causes cholestasis.
Primary sclerosing cholangitis
Primary sclerosing cholangitis (PSC) is a chronic, usually progressive, stricturing disease of the biliary tree. Remissions and relapses characterize the disease course. Primary sclerosing cholangitis may remain quiescent for long periods of time in some patients; in most cases, however, it is progressive.
The prevalence of primary sclerosing cholangitis in the United States is approximately 1–6 cases per 100,000 population. Most patients with primary sclerosing cholangitis are men (75%) with an average age of approximately 40 years at diagnosis. The overwhelming majority of patients affected with primary sclerosing cholangitis are Caucasian. The etiology is unknown but current opinion favors an immune cause. Management of this disease in the early stages involves the use of drugs to prevent disease progression. Endoscopic and surgical approaches are reserved for the time when symptoms develop. Liver transplantation may ultimately be required and offers the only chance for a complete cure. Patients with primary sclerosing cholangitis are at an increased risk for cholangiocarcinoma (10–15%).
Primary sclerosing cholangitis is a chronic fibrosing inflammatory process that results in the obliteration of the biliary tree and biliary cirrhosis. There is variability in the extent of involvement of the biliary system. The majority of patients with primary sclerosing cholangitis have underlying inflammatory bowel disease, namely ulcerative colitis or Crohn’s disease. Patients with primary sclerosing cholangitis are more likely to have ulcerative colitis than Crohn’s disease (85% versus 15%), with approximately 2.5–7.5% of all ulcerative colitis patients having primary sclerosing cholangitis. The strictures are located in both the intrahepatic and extrahepatic ducts in more than 80% of the patients, but about 10% of these patients have intrahepatic strictures only while less than 5% will have only extrahepatic strictures

Symptoms
Most patients with primary sclerosing cholangitis have no symptoms. These patients are usually diagnosed by the detection of abnormal biochemical tests of liver function on routine blood testing. Patients may remain asymptomatic for many years despite the presence of advanced disease.
When symptoms develop they are a result of obstruction to bile flow and include jaundice, itching, right upper quadrant abdominal pain, fever, and chills. Symptoms may also include weight loss and fatigue. The development of symptoms usually suggests the presence of advanced disease.
Causes
The cause of primary sclerosing cholangitis is not known. However there are several theories as to why damage to the bile duct occurs. Genetic abnormalities of the immune regulation, viral infection, toxins from intestinal bacteria, bacteria in the portal venous system, ischemic vascular damage, and toxic bile acids from intestinal bacteria are all factors that have been implicated in the pathogenesis of primary sclerosing cholangitis.
Currently genetic and immunological factors are most favored to be responsible for the damage to the bile ducts. This is because there is a familial occurrence of this disease and an association with HLA-B8, DR3, DR2, and DR4. Evidence of abnormal immuno-regulation is evidenced by infiltration of the bile ducts with lymphocytes, increased serum gamma globulins, increased circulating immune complexes, and increased metabolism of complement C3
References:
- Molecular pathogenesis of cholestasis – M.Trauner, P. Jansen, 2003
- Cholangiocyte biology. Tietz PS, Larusso NF (May 2006). Current Opinion in Gastroenterology 22 (3): 279–87.
- Cellular mechanisms of bile formation – M. Fuchs
- Secretion of Bile and the Role of Bile Acids In Digestion – R. Bowen
Some interesting articles:
- Bile formation: a concerted action of membrane transporters in hepatocytes and cholangiocytes. Zsembery et al., News Physiol. Sci. Volume 15 February 2000
- Molecular aspects of bile formation and cholestasis. Arrese and Trauner, TRENDS in molecular medicine, vol.9 no.12 december 2003
- Molecular mechanisms in bile formation. Meier and Steiger, News Physiol. Sci. Volume 15 April 2000
- Nutrition and bile formation. Tuchweber et al, Nutrition Research, Vol. 16, No. 6, pp. 1041-1080, 1996
Interesting videos:
http://www.youtube.com/watch?v=CCa3qcjGru0
http://www.youtube.com/watch?v=XjN8hUs1Wo0