INTRODUCTION

Cholesterol is the major constituent of the human brain, the most cholesterol-rich organ (containing about 20% if the body's total cholesterol).
Cholesterol is tightly regulated between brain cells (neurons, astrocytes, microglia and oligodendrocytes) and is essential for normal brain development. In fact cholesterol is required for synaptic and dendritic formation and for axonal guidance.
It is an important structural component of cellular membranes and myelin. The metabolism of brain cholesterol differs from that of other tissues, in fact is primarily derived by de novo synthesis and blood brain barrier prevents the uptake of lipoprotein from the circulation.
Cholesterol is synthesized via the isoprenoid biosynthetic pathway.
Isoprenoid biosynthesis starts with acetyl-CoA as a substrate, which by means of 6 subsequent enzyme reactions is converted into isopentenyl-pyrophosphate, the basic C5 isoprene unit used for synthesis of all subsequent isoprenoids. In total, at least 20 enzymes are involved for the generation of cholesterol.
The 3-hydroxy-3-methylglutaryl-coenzyme A reductase is the rate limiting enzyme in cholesterol biosynthesis and the target of statin pharmacotherapy.
The cholesterologenic pathway forms other important intermediates such as mevalonate, farnesyl pyrophosphate, squalene, and lanosterol.

There are two pools of cholesterol in the brain: one pool (containing up to 70%) consists of the myelin sheats of oligodendroglia, the other pool is made up by plasma membranes of astrocytes and neurons.
The majority of brain cholesterol accumulates between the perinatal period and adolescence when neurons are encircled by specialized plasma membranes termed myelin. After myelination, the metabolism of cholesterol in the adult brain is characterized by a very low turnover and minimal losses. However, recent results indicate that both cholesterol synthesis and degradation are active in the adult brain.
Cholesterol is vital to normal brain functions including learning and memory but that involvement is as complex as cholesterol synthesis, metabolism, and excretion. Excess cholesterol in the brain can lead to many signaling events via cholesterol metabolites, pro-inflammatory mediators, and antioxidant processes.
CHOLESTEROL SYNTHESIS INHIBITORS

Cholesterol synthesis inhibitors ( known as statins) are widely administered for reducing LDL cholesterol.
The main effect is the inhibition of HMG-CoA reductase, but an array of pleiotropic effects of statin therapy has been observed.
Statins differ in lipophilicity and studies with Simvastatin obtained the best results thanks to its capacity to pass blood-brain-barrier.
HMG-CoA reductase inhibitors act on the rate-limiting step in the pathway by which HMG-CoA is converted to mevalonate.
Statins reduce other by-products of the mevalonate pathway, including the isoprenoids farnesyl pyrophosphate and geranylgeranylpyrophosphate:
- FPP is also the substrate for production of coenzyme Q10 (CoQ10) and dolichol.
Coenzyme Q10 is an antioxidant, and dolichol may function as a radical scavenger. Of these two, only CoQ10 has been seriously studied in relation to statin treatment: statins decrease CoQ10 levels in plasma and tissue, which may be responsible for several of statins’ side effects.
- Besides this, farnesyl pyrophosphate and geranylgeranylpyrophosphate are also necessary for the post-translational lipid modification (prenylation) of several proteins that are tethered to the cell wall. These proteins have important roles in apoptosis, intracellular vesicular transport, cellular proliferation and differentiation, and the expression of additional membrane proteins (including cell adhesion molecules). Treatment with statins reduces prenylation and modifies several of these cellular functions, with the potential for therapeutic benefit in many neurodegenerative diseases.
Cholesterol: Its Regulation and Role in Central Nervous System Disorders, 2012
Now are described some therapeutic use of Statins in neurodegenerative diseases.
ALZHEIMER'S DISEASE
Alzheimer's disease is a degenerative disease characterized clinically by progressive memory loss and aberrant behavior. Usually it occurs late in life and without a known cause (sporadic Alzheimer's disease, SAD). Patients with this disease show a loss of synapses and neurons, and the formation of extracellular senile plaques and intracellular neurofibrillary tangles (NFTs).
Senile plaques consist largely of aggregated amyloid β-peptide (*Aβ*), which is liberated from the holoprotein, amyloid precursor protein (*APP*), by a sequential cleavages mediated by the β-secretase (Beta-site APP cleavage enzyme, BACE 1) and the γ-secretase complex. Intraneuronal accumulation of insoluble aggregates of tau in the form of NFTs is an other important hallmark of AD. Tau is a protein that normally stabilizes microtubules in axons, but in pathological conditions becomes hyperphosphorylated, so it is detached from microtubules and promotes the formation of insoluble tau aggregates.

The amyloid cascade hypotesis support the idea that amyloid β-peptide (Aβ) plays a central and even causative role for AD:
- Cleavage of β-amyloid precursor protein (APP) by BACE1 liberates its soluble ectodomain (sAPPβ) into the extracellular space.
- The resulting cell-associated COOH-terminal fragments, which can be either 99 or 89 amino acids in length, are subjected to intramembrane proteolysis mediated by γ-secretase, which generates a spectrum of Aβ peptides of varying length at the COOH terminus as well as the APP intracellular COOH-terminal domain (AICD). The predominant species of Aβ is 40 amino acids long (*Aβ40*), but the less abundant 42-amino acid variant (*Aβ42*) is more amyloidogenic and is the initial Aβ species that deposits into amyloid plaques in all forms of AD.
- Cleavage of APP within the Aβ sequence by α-secretase followed by γ-secretase cleavage, the non-amyloidogenic pathway, results in the production of a shorter, likely innocuous APP fragment (p3), along with the secreted ectodomain (sAPPα) and AICD.
Importantly, elevation of soluble Aβ oligomers strongly correlates with cognitive decline, consistent with the synaptotoxic properties exhibited by these peptides in various systems.
Linking Lipids to Alzheimer’s Disease: Cholesterol and Beyond, 2011

Statins have different effects on AD:
- AD brain displays a higher occurrence of “+adipose inclusions+” or “+lipoid granules+”, suggesting aberrant lipid metabolism. Subsequently, biochemical alterations of lipid composition have been reported in post mortem brain tissue derived from individuals with AD. However, an intimate link between lipid metabolism and AD was only established when the ε4 allele of the apolipoprotein E (APOE) gene was identified as the strongest genetic risk factor for SAD.
ApoE encodes a ~34 kDa protein that serves as a crucial regulator of cholesterol metabolism in the brain and triglyceride metabolism throughout the body. It mediates the uptake of lipoprotein particles in the brain via the low-density lipoprotein receptor related protein (LRP) and the very low-density family lipoprotein receptor. The role of ApoE4 in amyloid pathology is supported by evidence that it binds Aβ and modulates the aggregation and clearance of Aβ.

Between lipids, cholesterol has an important role.
(Simvastatin strongly reduces levels of Alzheimer's disease β-amyloid peptides Aβ42 and Aβ40 in vitro and in vivo,2001)
- Excess free cholesterol is coverted into cholesterol-esters by the enzyme acyl-coenzymeA cholesterol acyltransferase (*ACAT*). Increasing levels of this metabolite enhances Aβ release, while pharmacological inhibition of ACAT leads to the reduction of both Aβ and cholesterol-ester.
Many data suggest that the balance between free cholesterol and cholesterol esters is a key parameter controlling amyloidogenesis.

- Cholesterol can also directly modulate secretase activities leading to altered Aβ generation. Reducing membrane cholesterol levels decreases activity of both BACE1 and γ-secretase, leading to an additive reduction in Aβ generation. In fact cholesterol is a constituent of lipid-rafts, that have an important role in the amyloidogenic processing of APP.

APP and BACE1 are present in both raft and non-raft regions of the membrane but APP processing occurring within lipid rafts appears to be largely amyloidogenic, while outside lipid-rafts, APP is processed predominantly by the non-amyloidogenic, α-secretase, pathway.
Also core components of the γ-secretase complex, including presenilins, are also associated with lipid rafts. Accordingly, inhibition of γ-secretase activity leads to the accumulation of APP COOH-terminal products in lipid rafts. In contrast, other γ-secretase substrates, such as Notch 1 and Jagged 2, are largely processed in non-raft compartments. All this suggest that modulating the biophysical properties of rafts to decrease association of APP, BACE1 or presenilins with these lipid microdomains, may offer a therapeutic opportunity to reduce amyloidogenic processing of APP, thus delaying the progression of AD.
- An other important role of cholesterol inhibitor in AD is lead by HMG-CoA reductase pathway. This enzyme not only leads to the synthesis of cholesterol, but also provides eukaryotic cells with essential lipids, such as isoprenoids.

Thus, statins, which block HMG-CoA reductase, may also exert their effects through cholesterol-independent mechanisms. Short-chain isoprenoids farnesylpyrophosphate (*FPP*) and its metabolite geranylgeranylpyrophosphate (*GGPP*) are utilized for the isoprenylation of a wide variety of proteins, including small GTPases of the Ras, Rho and Rab families. Small GTPases act as molecular switches in a myriad of signaling and trafficking pathways and their isoprenylation, referred to as farnesylation and geranylgeranylation, allows for recruitment to the cytosolically-exposed leaflets of cellular membranes, where they exert their signaling actions. Recent evidence suggests that a perturbation of the metabolism of FPP and GGPP and thus GTPase signaling occurs in AD. GTPase signaling can control multiple aspects of amyloidogenesis, including the trafficking of APP, BACE1 and γ-secretase.

Recent studies have established that GGPP and to a lesser extent, FPP, are significantly elevated in the frontal cortex of AD patients, consistent with increased levels of their respective synthases. Importantly, pharmacological studies in various model systems have implicated short-chain isoprenoids as primary regulators of APP metabolism: while isoprenylation has been proposed to modulate the activity of γ-secretase in Aβ42 generation, statins were shown to promote ectodomain shedding of APP through the α-secretase pathway in a cholesterol-independent manner by inhibiting isoprenoid-mediated Rho/ROCK signaling, in favor of the non-amyloidogenic processing of APP. However, lowering isoprenoid levels, maintaining normal cholesterol levels, alters the traffic of APP leading to accumulation of β COOH-terminal fragments (βCTFs) and intracellular Aβ, suggesting that processing through the β-secretase pathway is enhanced.
All this indicate that isoprenylation differentially modulates all three secretase activities and there is pleiotropic effects of statins on APP metabolism by the modification of the cholesterol or the isoprenoid-specific processes.
Other studies (Cholesterol and Statins in Alzheimer’s Disease,2011)
demonstrate that when isoprenoid (but not cholesterol) levels are reduced by treatment with a statin in the presence of mevalonate, Aβ accumulates intracellularly. By contrast, when cholesterol is depleted by statin treatment in the absence of mevalonate , α-secretase processing of APP is enhanced, which in turn leads to less Aβ being secreted.
PAF-INDUCED DAMAGE
Platelet-activating-factor (PAF) is phosphoacylglycerol involved in a wide range of physiological and pathophysiological function.
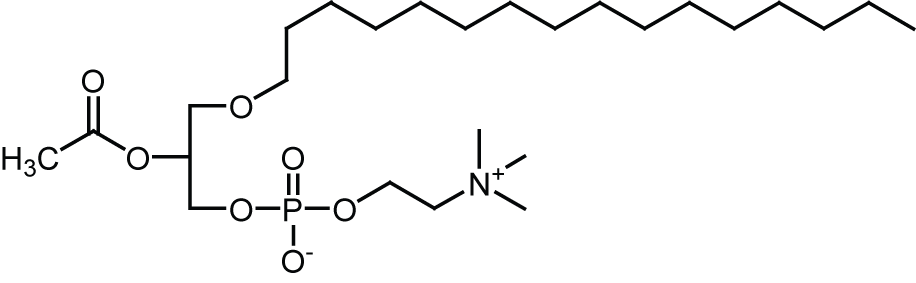
Although PAF plays roles in the normal functioning of neurons, higher concentrations are implicated in neuronal damage that accompanies ischemia, prion disease and Alzheimer' disease (AD).
The effects of PAF are mediated via a specific receptor with seven transmembrane spanning segments. The activation of downstream signalling pathways is dependent on interaction with pertussis toxin-sensitive G proteins.

Many experiments test the role of statin in this pathway and observate that pre-treatment with squalestatin and simvastatin protects neuron against PAF.
Cholesterol synthesis inhibitors protect against platelet-activating factor-induced neuronal damage,2007
This is not caused by the reduction of non-sterol products, such as isoprenoids that are essential for function of a wide variety of proteins, but by cholesterol-depletion.
In fact, if Simvastatin inhibits the enzyme HMG-CoA reductase causing a depletion of non-sterol products and cholesterol, squalestatin inhibits only squalene synthase thus reducing cholesterol production without affecting the production of non-sterol products.
This observation suggest that cholesterol depletion is responsible for neuroprotection. Furthermore, the protective effects of these drugs were reversed by the addition of squalene, a precursor of cholesterol synthesis that does not affect the production of non-sterol products. Besides this, the protective effects of squalestatin and simvastatin are not due to reduced amounts of PAF-receptor, but on the location within lipid-rafts.

In untreated neurons greater than 90% of the PAF receptors were found in lipid-rafts and this is very important because the activation of downstream signalling pathways by PAF is a dependent in interaction with pertussis toxin-sensitive G proteins, which also reside within lipid-rafts.
The formation of some lipid rafts is cholesterol-dependent and therefore susceptible to treatment with cholesterol synthesis inhibitors. After the treatment with this drugs, significantly less PAF-receptor was found within lipid rafts and more was found in the normal cell membrane. PAF-receptors outside lipid rafts fail to stimulate the G-proteins responsible for activation of downstream signalling pathways, that lead to neuronal death. As you can see on the image below, on point B, there is western-blot that show the amounts of PAF-receptors in lipid rafts/non-rafts from untreated neurons and neurons treated with squalestatin.

These results raise the possibility of using statins ad adjuvant therapy for neurodegenerative diseases in which PAF has been implicated, such as ischaemia, stroke and AD.
ANTIAPOPTOTIC PROTEINS PRODUCTION
Statins, especially Simvastatin, have another important role in therapies against neurodegenerative disease.
Simvastatin Stimulates Production of the Antiapoptotic Protein Bcl-2 via Endothelin-1 and NFATc3 in SH-SY5Y Cells,2010
In fact Simvastatins stimulate neuronal gene expression and protein levels of the more important antiapoptotic protein Bcl-2. These effects are indipendent from HMG-CoA reductase pathway.
Simvastatin can increase the levels of Endothelin-1 (*ET-1*), that normally increase Bcl-2 abundance via the transcription of the nuclear factor of activated rhymocytes (*NFATc*). This nuclear factor can be present in the cell in two different forms: one phosphorilated and one dephosphorilated. This last form can enter into the nucleus and bind Bcl-2 promoter.

ET-1, through the ET-1 receptor, leads to the rise of calcium level in the cell, and to the activation of a phosphatase-calcium dependent (Ca2+ /Calcineurin)resulting in dephosphorylation of NFATc and allowing it to translocate to the nucleus and bind to consensus sequences on the Bcl-2 promoter.
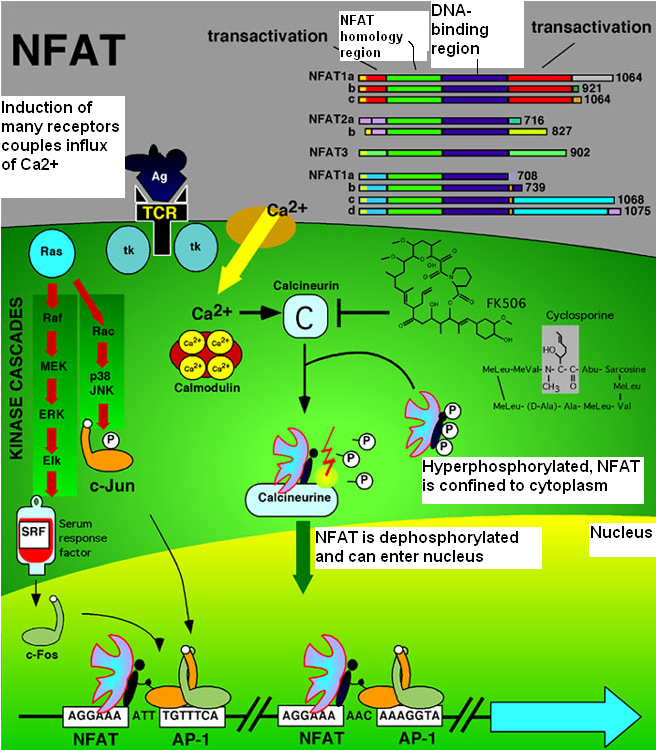
Simvastatin are demonstrated to increase both intracellular and secreted ET-1 protein levels. These drugs increase also the traslocation of NFATc (in the isoform 3), in the nucleus. Simvastatins have the opposite effect on NFATc1 and did not alter levels of NFATc4 in the nucleus
NFATc proteins typically cannot bind DNA alone due to their weak binding to DNA and require a cotranscription binding protein, collectively termed NFATn.
NFATn proteins are a diverse group of proteins and include, for example, AP-1, GATA, cMAF, and MEF2 family members. The most well-known NFATc cotranscription factor is the AP-1 complex of c-Fos and c-Jun.
This effect of Simvastatin will provide a new approach to statin-induced neuroprotection.

EPILEPSY
Epilepsy affects 50 million people worldwide. About 30% of epileptic patients have temporal lobe epilepsy (TLE). TLE, which eventually leads to cognitive deficits, can not be controlled by antiepileptic drugs or surgical removal of epileptic focus.
Administration of Simvastatin after Kainic Acid-Induced Status Epilepticus Restrains Chronic Temporal Lobe Epilepsy,2011
Simvastatin, thanks to its high blood-brain-barrier, is the most effective statin against kainate-induced excitotoxicity and memory impairment.

This is related to the role of statin in regulating NMDA receptors and in anti-inflammation effect:
- Simvastatins also inhibit neuroinflamation. Pro-inflammatory cytokines play key roles in the epileptogenic cascade, including seizure-related pathological changes in hippocampus, such as neuronal death and reactive gliosis. KA-induced epilepsy injury is accompanied by upregulation of cytokines including TNF-α and IL-1 in glial cells. Manipulation of level of cytokines in inflammatory cascades could have a potential effect on KA-induced injury.
Simvastatin decreases levels of IL-1 β and TNF-α by regulating nuclear factor-kB transcription pathway and decreasing isoprenylation protein involved in cellurar signaling.
Inflammatory Markers and the Metabolic Syndrome: Insights From Therapeutic Interventions,2005

Thanks to this, Simvastatin-mediated early suppression of the inflammatory cascade may result in a marked reduction in secondary neuronal damage after status epilepticus.
- Therefore, the neuroprotective effect of simvastatin may be also related to increased BDNF level that prevents neurons from lesion-induced degeneration.

Thus, both anti-inflammation and neurotrophy effect of simvastatin may contribute to its neuroprotective role found.
- At last, Simvastatins suppress astrocytosis, that is a source of cytokines including IL-1β, TNF-α, and IL-6 and is associated with the chronic relapsing forms. Astrocytosis may exacerbate inflammation by inducing the migration of other leukocytes into the injured site, interrupting blood-brain-barrier function and producing reactive oxygen species and cytotoxic edema,
The simvastatin-mediated suppression of astrocytosis may contribute to inhibition of neuroinflammation and neuronal loss, thus exert a neuroprotective role in KA-induced injury.
PARKINSON'S DISEASE
Parkinson's disease (PD) is the second most common neurodegenerative disorder and is characterized by disturbance of the central dopaminergic system and imbalances in some non-dopaminergic systems, inclunding the glutamatergic system.

- Animal and clinical evdence has shown that statin have effects on PD, and this is mediated through NMDA receptors, that are significantly reduced in patients affected by PD due to 6-hydroxydopamine (*6-OHDA*) neurotoxicity.

Simvastatin Prevents Dopaminergic Neurodegeneration in Experimental Parkinsonian Models: The Association with Anti-Inflammatory Responses,2011
Simvastatin has shown to affect D1/D2 receptors, alter dopamine content in various brain regions and up-regulate the NMDA receptors in different regions of the brain.
- Simvastatin also regulates the inflammatory responses, characterized by activation of microglia and accumulation of inflammatory mediators such as cytokines and proteases, and activates the neurotrophic factor BDNF.
Thanks to these evidences Statins can be associated with anti-inflammatory and anti-excitotoxicity effects.
OTHER FIELDS OF INTEREST
Other possible applications of Statins are cerebrovascular disease, such as acute ischemic stroke, intracerebral hemorrhage and subarachnoid hemorrhage.
3-Hydroxy-3-methylglutaryl–Coenzyme A Reductase Inhibitors in the Treatment of Central Nervous System Diseases,2010
CONCLUSION
At last, statins can have a double effect on the brain, because they have both a neuroprotective and a neurotoxic effect.

Statins have been proposed in the treatment of multiple central nervous system diseases, but the usefulness of this drugs in inflammatory and degenerative disease of the central nervous system needs to be tested too.
In fact an great number of studies focused their attention on these drugs, many times with different conclusions. Statins rapresent a possible drug for many neuronal diseases but more studies to test their positive and negative effects are required.