ACHONDROPLASIA
DEFINITION
Achondroplasia, is a common cause of dwarfism. It occurs as a sporadic mutation in approximately 75% of cases (associated with advanced paternal age) or may be inherited as an autosomal dominant genetic disorder.It is caused by a change in the DNA for fibroblast growth factor receptor 3 (FGFR3), which causes an abnormality of cartilage formation. If both parents of a child have achondroplasia, and both parents pass on the mutant gene, then it is very unlikely that the homozygous child will live past a few months of its life. More than 250,000 people in the world are diagnosed with Achondroplasia currently. Achondroplasia diagnosis occurs somewhere between one in every 10,000 to one in every 30,000 live births.
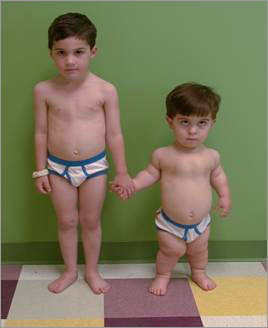
An example of difference between a normal child and an acondroplastic child is illustrated in the figure bellow:
GENETIC CAUSES
Achondroplasia is caused by a mutation, or change, in the fibroblast growth factor receptor 3 gene (FGFR3) located on the short arm of chromosome 4. FGFR3 is one of four high affinity FGF receptors (FGFR1–4) [11–13]. It functions as transmembrane receptor tyrosine kinase possessing an extracellular ligand-binding domain, a transmembrane domain and an intracellular split tyrosine kinase domain . In a normal (non-mutated) gene, base number 1138 is guanine (G). In most individuals with achondroplasia (98%), this guanine (G) has been replaced with adenine (A). In a small number of individuals with achondroplasia (1%), this guanine (G) has been replaced with cytosine ( C ). Both of these small substitutions cause a change in the fibroblast growth factor receptor (FGFR) (change Glycin to Arginin) that affects the function of this receptor.
The penetrance of the achondroplasia mutation is 100%. The vast majority of infants with FGFR3 mutations are born to parents without FGFR3 mutations, although there is a strong correlation with advanced paternal age (over 35 years). These findings were initially attributed to increased mutability of FGFR3 during spermatogenesis. However, recent observations, including the detection of FGFR3 in all germ cells except for elongated spermatids in adult men and failure to detect sufficiently high mutation rates in sperm from older males, have led to the alternative explanation that sperm bearing mutant FGFR3 have a selective advantage over sperm bearing normal FGFR3receptor.
In normal conditions the receptor after it has interacted with his ligand (fibroblast growth factor) it activates and acts a negative control on the growth of cartillage cells that forms bones. The mutation of the gene induce a constant attivation of the receptor which constantely inibits the growing up of the bones cartilage.

Figure 1. Domain structure of FGFR3 and major sites of mutations. Ig: immunoglobulin, AB: acid box, TM: transmembrane, TKp/d: proximal and distal tyrosine kinase domains, ACH: achondroplasia, HYP: hypochondroplasia, TD: thanatophoric dysplasia, SADDAN: severe achondroplasia with developmental delay and acanthosis nigricans.
http://www.gghjournal.com/volume22/4/featureArticle.cfm
MOLECULAR PATHOGENESIS
Like other FGFRs, FGFR3 is activated by FGF ligands released from cells near the FGFR3-bearing target cells. The binding of FGF ligands to FGFR3 monomers leads to receptor dimerization. Which of the 22 known FGFs is (are) the physiologic ligand(s) for FGFR3 is (are) not known, although FGFs 2, 4, 9 and 18 are probably the best candidates based on the distribution of expression and ability to bind and activate FGFR3 in in vitro assays.It is also conceivable that different FGF ligands activate FGFR3 in different physiologic situations. Heparin sulfate-bearing proteoglycans on the cell surface, such as syndecans, as well as alternative splicing of ligand-binding subdomains, influence binding specificity.
Dimerization activates the intrinsic tyrosine kinase activity of the receptor and promotes transphosphorylation of key tyrosine residues in the cytoplasmic domain. These residues serve as docking sites for adapter proteins and signal effectors that are recruited to the activated receptors and which propagate FGFR3 signals.
http://www.gghjournal.com/volume22/4/featureArticle.cfm
SIGNALING PATHWAY
FGFR3 signals influence a variety of cellular events and processes largely through inducing or repressing expression of target genes in a cell-specific context. Four main signaling pathways have been identified to date to propagate FGFR3 signals: STAT , MAPK , PLC- g , and PI3K-AKT (signal transducer and activator of transcription 1, mitogen-activated protein kinase, phospholipase C gamma, phosphatidylinositol phosphate-3-kinase-serine/threonine kinase [protein kinase B]) with the first 2 receiving the most attention.
• STAT1 signals are thought to induce expression of mitotic inhibitors, such as the cdk inhibitor p21 . Using microarrays to assess changes in gene expression in chondrocytic cells, Dailey et al showed that FGFs initiate signals in multiple pathways that result in the induction of antiproliferative functions and down regulation of growth-promoting molecules.
• Two MAPK pathways have been implicated, the strongest evidence coming from transgenic mice in whom expression of constitutively active members of the 2 pathways was targeted to cartilage, including growth plate cartilage. Expression of activated MKK6 , which specifically activates the MAPK-p38 pathway, inhibits chondrocyte proliferation in part through induction of the transcription factor Sox 9 . Chondrocyte hypertrophy was also inhibited in these dwarf mice. Expression of constitutively active MEK1, which specifically activates the MAPK-ERK pathway, produced a similar dwarf phenotype, but through inhibition of terminal chondrocyte differentiation with no inhibitory effect on cell proliferation. These observations underscore the importance of both chondrocyte proliferation and terminal (hypertrophic) differentiation in linear bone growth and the central role of FGFR3 in negatively regulating these events.
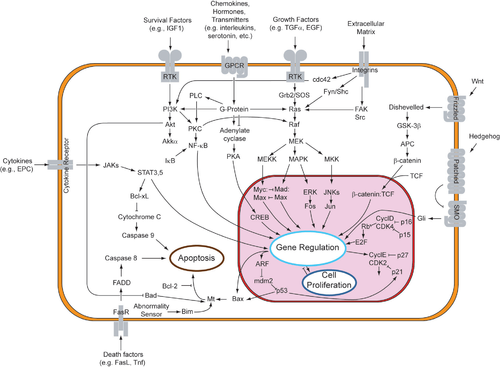
It is important to emphasize that FGFR3 is one of many physiologic regulators that modulate linear bone growth. Its normal function is as a negative regulator. The mutations associated with achondroplasia and related conditions are thought to act through exaggeration or enhancement of this normal physiologic function rather than through acquisition of new functions.
http://www.gghjournal.com/volume22/4/featureArticle.cfm
http://academic.regis.edu/jbetz/BL610Bclass/Achondroplasiareview.pdf
DIAGNOSIS / CLINICAL CHARACTERISTICS
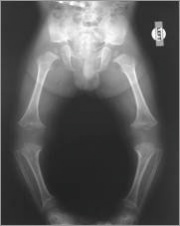
At birth or during infancy, achondroplasia is generally diagnosed with x-rays and a physical examination. If there is any question about the diagnosis, genetic testing using a blood sample can be done to look for a mutation (change) in the gene that causes achondroplasia.
Before birth, achondroplasia may be suspected in the fetus if an ultrasound shows shortened bones and other bone abnormalities. In such cases, the health care provider may recommend amniocentesis to confirm the diagnosis. A child with achondroplasia has a relatively normal torso and short arms and legs. The upper arms and thighs are more shortened than the forearms and lower legs. Generally, the head is large, the forehead is prominent and the nose is flat at the bridge. Sometimes, the large head size reflects hydrocephalus (excess fluid in the brain) and requires surgery. Hands are short with stubby fingers. There is a separation between the middle and ring fingers (trident hand). Feet are generally short, broad and flat. Most individuals with achondroplasia eventually reach an adult height of about 4 feet .
The average height of an adult male with achondroplasia is 131 centimeters (52 inches, or 4 feet 4 inches), and the average height of an adult female with achondroplasia is 124 centimeters (49 inches, or 4 feet 1 inch).
http://www.marchofdimes.com/baby/birthdefects_achondroplasia.html
http://lifestyle.iloveindia.com/lounge/achondroplasia-symptoms-8946.html
TREATMENT
Health care providers closely monitor the growth and development of children with achondroplasia. Though there is currently no way to normalize skeletal development of children with the disorder, most complications can be effectively treated.
http://www.marchofdimes.com/baby/birthdefects_achondroplasia.html
However a number of potential therapeutic strategies have emerged such as:
- The most attention in achondroplasia has gone to inhibiting the FGFR3 tyrosine kinase through small chemical inhibitors. Selective FGFR3 kinase inhibitors have been developed and show promise in cell and organ culture experiments, but to date none has shown success in whole animals.
- An alternative approach has involved generating antibodies to block FGFR3 activation. Although highly specific humanized antibodies have been developed, there have been no reports to date of success beyond cell culture experiments in which they block receptor activation well.
- The therapeutic use of CNP or a CNP analog that could activate NPR-B signaling pathway to counter excessive FGFR3 signals transmitted through the MAPK–ERK and possibly MAPK-p38 pathways has been proposed. This approach is appealing because other natriuretic peptides have been used clinically for their hemodynamic effects in adults and even in children. While they appear to be safe at least in the short term, a major drawback is their very short half-life requiring them to be administered by infusion, which would not be satisfactory for long-term treatment of achondroplasia.

Figure 2. Signaling pathways and potential therapeutic strategies. FGFR3 signals are propagated through STAT1, MAPK-ERK, MAPK-p38 and probably other pathways which inhibit growth plate chondrocyte proliferation, post-mitotic matrix synthesis and terminal (hypertrophic) differentiation. The CNP-NPR-B pathway inhibits the MAPK pathways. Proposed therapeutic strategies include chemical inhibition of FGFR3 tyrosine kinase, antibody blockade of ligand-induced receptor activation, and enhancement of CNP-NPR-B signals.
http://tratando-acondroplasia.blogspot.it/2012/02/first-potential-pharmacological-therapy.html
http://www.gghjournal.com/volume22/4/featureArticle.cfm
CNP prevented the shortening of achondroplastic bones by correcting the decreased intracellular Matrix syntesis in the growth platethrough inhibition of the MAPK pathway of FGF signaling
CONCLUSION
The tyrosine kinase-mediated transmembrane receptor FGFR3 is an important negative regulator of linear bone growth acting mainly through the STAT1, MAPK-p38, and MAPK-ERK signaling pathways to inhibit chondrocyte proliferation and terminal differentiation in the growth plate. Mutations that enhance these actions produce the qualitative achondroplasia clinical phenotype; the extent of this enhancement correlates with the severity of this phenotype. The mutations act through promoting or stabilizing the dimerization required for receptor activation, by directly activating kinase activity through conformational change of the receptor and by slowing of receptor degradation. Several strategies have been proposed to therapeutically counter the increased FGFR3 signal output, including chemical tyrosine kinase inhibitors and blocking antibodies, both selective for FGFR3 and activation of the CNP-NPR-B-cGMP pathway, which antagonizes MAPK-ERK/p38 signals downstream of FGFR3. All 3 strategies have shown success in cell and organ culture systems, but not yet in whole animal trials, perhaps because they may need to be targeted directly to growth plate chondrocytes to achieve therapeutic effect restricted to growing bones.