Introduction
The pyroptosis is a form of cell death intermediate between apoptosis and necrosis; this pathway of cell death is uniquely dependent on caspase-1. Caspase-1 is not involved in apoptotic cell death and caspase-1-deficient cells respond normally to most apoptotic signals. Caspase-1 is activated during pyroptosis by a large supramolecular complex termed the pyroptosome (also known as an inflammasome). An important function of caspase-1 is to process the proforms of the inflammatory cytokines, IL-1β and IL-18, to their active forms. The mechanism and outcome of this form of cell death are distinctly different from these aspects of apoptosis, which actively inhibits inflammation. In stead of apoptosis, pyroptosis consists in cellular swelling, plasma membrane rupture and release of intracellular content into the extracellular milieu, including cytosolic enzymes like lactate dehydrogenase (LDH). LDH release was readily detected after HIV infection.
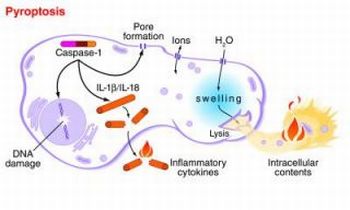
The term pyroptosis was proposed from the Greek roots “pyro,” relating to fire or fever, and “ptosis”, denoting falling, to describe proinflammatory programmed cell death. The observed caspase-1 activation or dependence during cell death in the immune, central nervous, and cardiovascular systems indicates that pyroptosis plays a significant role in a variety of biological systems.
CD4 T-cell death by HIV-1 is mediated by pyroptosis
The progressive loss of CD4 T cells in HIV-infected individuals contributes to the rise of AIDS. Particularly mysterious has been the observation that HIV-1 infection results not only in the death of activated, productively infected CD4+ T cells (those in which the virus successfully replicates) but also in ‘bystander’ CD4+ T cells that do not seem to be infected. HIV-1 replication in productively infected CD4+ T cells kills them quickly, within one to two days. This direct killing is apparent during acute infection, when virus levels are high and massive depletion of CD4+ T cells occurs in the gastrointestinal tract. However, in the absence of treatment, most of the CD4+ T-cell loss associated with the infection occurs during the prolonged asymptomatic phase between the acute stage and the development of AIDS. During this period, the number of activated, productively infected CD4+ T cells is low, suggesting that the infection may promote the death of quiescent (non-activated) cells.
Levels of immune activation are high in untreated HIV-1 infection, perhaps reflecting the translocation of microbial products across a compromised gastrointestinal barrier, and it is commonly assumed that this immune activation is responsible for CD4+ T-cell loss. In a recently study (Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection, Nature 2014), using cultures of human cells isolated from the spleen or tonsils, Doitsh and his collegues demonstrate that more than 95% of CD4+ T cells that die following HIV-1 infection are quiescent cells that undergo pyroptosis. Only a small proportion of the dying cells were activated, productively infected CD4+ T cells undergoing apoptosis.

The HIV-1 resistance of bystander CD4 T cells arises in part from a host cell deoxynucleoside triphosphate (dNTP) triphosphohydrolase [called sterile alpha motif (SAM) domain and histidine–aspartate (HD) domain–containing protein 1 (SAMHD1)]. This enzyme depletes cytosolic dNTP pools, thus depriving HIV-1 of essential building blocks. Ironically, the incomplete viral DNA genomes produced as a consequence of SAMHD1’s protective inhibition of HIV-1 now appear to initiate pyroptosis as well as the elaboration of the inflammatory cytokine interleukin-1β (IL- 1β), which lures even more CD4 T cells into the killing zone.
In another recently study (IFI16 DNA Sensor Is Required for Death of Lymphoid CD4 T Cells Abortively Infected with HIV, Science 2014) Monroe and others ,using unbiased proteomic and targeted biochemical approaches, as well as two independent methods of lentiviral short hairpin RNA–mediated gene knockdown in primary CD4 T cells, identified interferon-γ–inducible protein 16 (IFI16) as a host DNA sensor required for CD4 T cell death due to abortive HIV infection. IFI16 initiates an innate immune response that, rather than protecting the host, drives the debilitating CD4 T cell depletion that underlies progression to AIDS in untreated HIV-infected individuals.
The cycle of abortive infection, inflammatory death, and recruitment of new cells likely explains how this innate host response is undermined and, in fact, centrally contributes to HIV pathogenesis. IFI16 has been identified as a critical DNA sensor required for cell death during abortive HIV-1 infection.
These results suggest that abortive infection of resting CD4+ T cells triggers pyroptosis and thus contributes to two major drivers of HIV 1 pathogenesis: the depletion of CD4+ T cells and systemic inflammation.
The studies by Doitsh et al. and Monroe et al. may set the stage for a new class of anti–HIV-1 therapeutics.
New possible Therapies
The role of caspase 1 in the chronic inflammatory response has attracted therapeutic interest.
VX-765 is a caspase 1 inhibitor that has been tested in chronic epilepsy and psoriasis, and found in a phase IIa trial to be safe and well tolerated over the six-week length of the trial. VX-765 also blocked caspase 1 cleavage, IL-1β secretion and CD4 T-cell death in HIV-infected tonsillar and splenic Human Lymphoid Aggregate Cultures. Cell death was not markedly inhibited by VRT-043198 (the active form of the VX765 pro-drug), probably because of reduced cellular permeability.

HIV-1 infection was not restored to productive infection when caspase 1 was blocked. These findings demonstrate that a small-molecule inhibitor of caspase 1, shown to be safe in humans, suppresses CD4 T-cell death and inflammation elicited in lymphoid tissues by HIV-1.
Conclusions
Pyroptosis probably promotes the rapid clearance of various bacterial infections by removing intracellular replication niches and enhancing the host’s defensive responses through the release of pro-inflammatory cytokines and endogenous danger signals.
However, in pathogenic chronic inflammation, such as in HIV infection, pyroptosis is not a protective response and does not lead to clearance of the primary infection. In fact, pyroptosis appears to create a pathogenic vicious cycle in which dying CD4 T cells release inflammatory signals that attract more cells into the infected lymphoid tissue to die and to produce more inflammation. These events establish a chronic state of inflammation that probably fuels disease progression and tissue injury (The Journal of Clinical Investigation, Volume 121, Issue 3).
Chronic inflammation might also promote maintenance of the latent HIV reservoir through the deregulated action of the IL-7 or IL-15 cytokines stimulating homeostatic proliferation of memory CD4 T cells. In this regard, it will be interesting to assess to what extent pyroptosis persists in lymphoid tissues of HIV-infected subjects on effective anti-retroviral therapy. The depletion of CD4 T cells and the development of chronic inflammation are signature processes in HIV pathogenesis that propel disease progression. Doitsh’s studies reveal how pyroptosis provides an unexpected link between these two disease-promoting processes. The pathogenic cycle of cell death and inflammation created by pyroptosis obligately requires the activation of caspase 1. As such, it may be possible to break this pathogenic cycle with safe and effective caspase 1 inhibitors. These agents could form a new and exciting ‘anti-AIDS’ therapy for HIV-infected subjects in which the treatment targets the host instead of the virus.