DEFINITION
Roberts syndrome (RBS; OMIM#268300), also known as Roberts-SC phocomelia syndrome (OMIM#269000), Pseudothalidomide Syndrome, Roberts-SC (Pseudothalidomide), SC-Phocomelia Syndrome is a rare genetic disorder characterized by growth delays before and after birth (pre- and postnatal growth deficiency ranging from mild to severe); malformations of the arms and legs (limbs); distinctive abnormalities of the skull and facial (craniofacial) region. Severely affected infants may be stillborn or die shortly after birth.
A condition called SC phocomelia syndrome was originally thought to be distinct from Roberts syndrome; however, it is now considered to be a mild variant. "SC" represents the first letters of the surnames of the two families first diagnosed with this disorder.
GeneReviews® 2009
It is caused by mutations in the ESCO2 (establishment of sister chromatid cohesion N-acetyltransferase 2 also known as establishment of cohesion 1 homolog 2 or ECO1 homolog 2), gene located on the short arm (p) of 8th chromosome (8p21.1), the only known gene responsible for this disease.
Roberts Syndrome
EPIDEMIOLOGY
Named after John B. Roberts, who first described the syndrome in 1919, it is one of the rarest autosomal recessive disorders, affecting approximately 150 known individuals.
The Syndrome is both autosomal, in that there are equal numbers of copies of the gene in both males and females, and recessive, meaning the child must inherit the defective gene from both parents.
Roberts syndrome can affect both males and females. Although the disorder is rare, the affected group is diverse. Approximately 150 individuals of diverse racial and ethnic backgrounds have been reported. Some cases of Roberts syndrome have had parents who were related by blood ( consanguineous ). All individuals carry a few abnormal genes. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
Wide clinical variability is observed among affected individuals, including siblings.
The prognosis is relatively unfavorable, for an individual with RBS depends on the malformations present: the severity of manifestations correlates with survival.
The mortality rate is high among severely affected pregnancies, in the newborn period or early childhood, due to cardiac or renal malformations; however, mildly affected individuals may survive to adulthood. The cause of death has not been reported for most affected individuals; in five cases it was reported to be infection.
The SC phocomelia and the Roberts syndrome: nosologic aspects 1977
Discovery of the syndrome
The discovery of ESCO2 as the gene responsible for Roberts syndrome was made by studying samples from fifteen families affected by Roberts syndrome. In 1995, Vega and Gordillo, two Colombian geneticists, set out to fully understand Roberts syndrome, since they noticed an unusually high number of Roberts syndrome patients at the Universidad Nacional de Colombia. The two Colombian geneticists tracked down a total of seven families with Roberts syndrome and discovered that four out of the seven families shared a common 18th century ancestor. Using this information, Vega and Gordillo were able to pinpoint the gene responsible for Roberts syndrome, which was ESCO2.
Fifteen Year Hunt Uncovers Gene Behind 'Pseudothalidomide' Syndrome.
SYMPTOMS

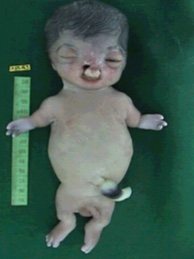
The symptoms associated with Roberts syndrome vary widely from case to case even among members of the same family.
Growth retardation of prenatal onset is the most consistent finding in all affected individuals. Postnatal growth retardation can be moderate to severe and correlates with the severity of the limb and craniofacial malformations.
Limb malformations include symmetric mesomelic shortening and anterior-posterior axis involvement in which the frequency and degree of involvement of long bones is, in decreasing order: radii, ulnae, and humeri in the upper limbs; fibulae, tibiae, and femur in the lower limbs. The degree of limb abnormalities follows a cephalo-caudal pattern: the upper limbs are more severely affected than the lower, with several cases of only upper limb malformations.
Hand malformations include brachydactyly and oligodactyly. The thumb is most often affected by proximal positioning or digitalization, hypoplasia, or agenesis. The fifth finger is the next most affected digit with clinodactyly, hypoplasia, or agenesis. In severe cases, only three fingers are present (and rarely, only one finger).
Craniofacial abnormalities include: cleft lip and/or cleft palate, premaxillary prominence, micrognathia, microbrachycephaly, midfacial capillary hemangioma, malar flattening, downslanted palpebral fissures, widely spaced eyes, exophthalmos resulting from shallow orbits, corneal clouding, underdeveloped ala nasi, beaked nose, and ear malformations. Mildly affected individuals have no palatal abnormalities or only a high-arched palate. The most severely affected individuals have fronto-ethmoid-nasal-maxillary encephalocele.
Correlation between the degree of limb and facial involvement is observed: individuals with mild limb abnormalities also have mild craniofacial malformations, while those with severely affected limbs present with extensive craniofacial abnormalities.
Other abnormalities may be observed:
Heart. Atrial septal defect, ventricular septal defect, patent ductus arteriosus
Kidneys. Polycystic kidney, horseshoe kidney
Intellectual ability. Intellectual disability is present in the majority of affected individuals; however, normal intellectual and social development has been reported
Premature centromere splitting in a presumptive mild form of Roberts syndrome 1984
Incidental detection of premature centromere separation in amniocytes associated with a mild form of Roberts syndrome 1988
Roberts syndrome: phenotypic variation, cytogenetic definition and heterozygote detection 1991
Roberts/pseudothalidomide syndrome and normal intelligence: approaches to diagnosis and management 1992
DIAGNOSIS
A diagnosis of Roberts syndrome is suspected based upon a thorough clinical evaluation, detailed patient history and identification of characteristic abnormalities. A diagnosis may be confirmed by chromosomal analysis that detects characteristic premature centromere separation (puffing) on various chromosomes. Absence of puffing does not exclude the diagnosis, as it was reported to be absent in some clinically diagnosed cases.
In some cases, it is possible that a diagnosis of Roberts syndrome may be suspected before birth (prenatally) based upon specialized tests, such as amniocentesis (usually performed at ~15-18 weeks’ gestation), chorionic villus sampling (CVS, usually performed at ~10-12 weeks’ gestation), or ultrasonography. Chromosomal studies performed on such fluid or tissue samples may reveal premature centromere separation (puffing) in mitotic cells. During fetal ultrasonography, reflected sound waves create an image of the developing fetus, potentially revealing certain developmental abnormalities suggestive Roberts syndrome (e.g., limb abnormalities).

The diagnosis of RBS is confirmed by molecular testing for ESCO2 mutations. The presence of mutations in this gene is strictly correlated with the centromere puffing phenomenon.
Testing
The diagnosis of RBS relies on cytogenetic testing in peripheral blood of individuals with suggestive clinical findings. Standard cytogenetic preparations stained with Giemsa or C-banding techniques show the characteristic chromosomal abnormality of premature centromere separation (PCS) and separation of the heterochromatic regions (also termed heterochromatin repulsion [HR]) in most chromosomes in all metaphases (Figure 1).

C-banding of metaphase chromosomes. Arrows show selected chromosomes with premature centromere separation. Large arrow head points to 'splitting' of the Y chromosome heterochromatic region. Open arrows show selected chromosomes with normal C-banded.
The centromere and the heterochromatin are affected in RBS.)
The cytogenetic abnormalities is observed in standard cytogenetic preparations and the prematurely separating centromeres during metaphase rather than in anaphase. PCS is related to the most probable pathologic mechanism and associated spindle checkpoint activation and impaired cell proliferation.
Many chromosomes display a "railroad track" appearance as a result of the absence of the primary constriction and presence of "puffing" or "repulsion" at the heterochromatic regions around the centromeres and nucleolar organizers.
The heterochromatic region of the long arm of the Y chromosome is often widely separated in metaphase spreads.
Aneuploidy, micronucleation, and multilobulated nuclei are also common findings in RBS cell cultures.
Carrier status cannot be determined by cytogenetic analysis.
Management
Treatment of manifestations: Individualized treatment aimed to improve quality of life; surgery for cleft lip and/or palate, for correction of limb abnormalities, and to improve proper development of the prehensile hand grasp. Prostheses, speech assessment and therapy, aggressive treatment of otitis media, special education for developmental delays, and standard treatment for ophthalmologic, cardiac, and renal abnormalities may be indicated.
Surveillance: Periodic follow up of psychomotor development and physical growth; follow-up assessment of speech development and hearing if cleft lip and palate are present; screening for developmental delays or learning disorders; monitoring for specific ophthalmologic, cardiac, or renal anomalies.
Prenatal testing for pregnancies at increased risk is possible by a combination of ultrasound examination and cytogenetic testing, or by molecular genetic testing if the mutations have been identified in the family.
Roberts Syndrome 2006
Molecular Genetic Testing

The ESCO2 gene has a specific effect on cell division in Roberts syndrome patients. In normal cell division, each chromosome is copied and then attached to its newly formed copy at the centromere. However, in Roberts syndrome cell division, the copies are frequently not attached at the centromere. As a result, the chromosomes do not get lined up properly, which causes the cell to divide very slowly or even to not divide at all. The new cells typically will have too many or too few chromosomes. The odd number of chromosomes causes the defective cells to die, which leads to the malformations associated with Roberts syndrome.
It is widely expressed in fetal tissues, while, in adult, it is expressed in thymus, placenta and small intestine. It is a protein-coding gene acetyltransferase, required for the establishment of sister chromatid cohesion, couples the processes of cohesion and DNA replication to ensure that only sister chromatids become paired together. In contrast to the structural cohesins, the deposition and establishment factors are required only during the S phase. Acetylates the cohesin component SMC3
RBS chromosomes have a lack of cohesion involving the heterochromatic C-banding regions around centromeres and the distal portion of the long arm of the Y chromosome (known as premature centromere separation, heterochromatin repulsion or puffing, or RS effect).
Cellular alterations in RBS include:
1. lack of cohesion at the heterochromatic regions around centromeres and the long arm of the Y chromosome,
2. reduced growth capacity,
3. hypersensitivity to DNA damaging agents.
The following table shows the features of ESCO2.
| Gene Symbol | Chromosomal Locus | Protein Name |
| ESCO2 | 8p21.1 | N-acetyltransferase ESCO2 |
Normal allelic variants ESCO2 comprises 11 exons distributed over 30.3 kb, transcribed into a 3,376 nucleotide mRNA with an open reading frame of 1,806 nucleotides.
The normal variant p.Ala80Val in exon 3 has a heterozygosity of 0.235, but its functional significance is not known.
Pathogenic allelic variants The mutations in ESCO2 are highly variable. Different mutations in ESCO2 have been reported in families with Roberts syndrome (RBS) ( Table 2 ). All but two are frameshift or nonsense mutations that lead to protein truncation or nonsense-mediated decay. The two missense mutations lead to amino acid substitutions of highly conserved amino acids in the acetyltransferase domain. Exonic or whole gene deletions or duplications have not been reported.
Table 2
| Class of Variant Allele | DNA Nucleotide Change | Protein Amino Acid Change |
| Normal | c.239C>T | p.Ala80Val |
| c.252_253delAT | p.Ser85PhefsTer6 (p.V84fs*7) |
| c.294_297delGAGA | p.Arg99SerfsTer2 (p.E98fs*2) |
| c.307_311delAAAGA (c.307_311delAGAAA) | p.Lys103GlufsTer2 (p.I102fs*1) |
| c.308_309delAA | p.Lys103ArgfsTer3 (p.K103fs*3) |
| c.417dupA (c.411_412insA) | p.Pro140ThrfsTer8 (p.K138fs*10) |
| c.505C>T | p.Arg169Ter |
| c.604C>T | p.Gln202Ter |
| c.745_746delGT | p.Val249GlnfsTer1 |
| c.751dupG (c.750_751insG) | p.Glu251GlyfsTer30 (p.E251fs*30) |
| c.760dupA (c.751_752insA) | p.Thr254AsnfsTer27 (p.K253fs*26) |
| c.760delA (c.752delA) | p.Thr254LeufsTer13 (p.K253fs*12) |
| c.764_765delTT | p.Phe255CysfsTer25 (p.F255fs*25) |
| c.875_878delACAG | p.Asp292GlufsTer48 (p.D292fs*47) |
| Pathogenic | c.879_880delAG (c.877_888delAG) | p.Arg293SerfsTer7 (p.R293fs*7) |
| c.955+2_+5delTAAG | -- |
| c.1111dupA (c.1104_1105insA) | p.Thr371AsnfsTer32 (p.K369fs*34) |
| c.1111_1112insG0 | p.Thr371SerfsTer32 (p.T371fs*32 |
| c.1131+1G>A | -- |
| c.1132-7A>G | -- |
| c.1263+1G>C | -- |
| c.1269G>A | p.Trp423Ter |
| c.1354-18G>A | -- |
| c.1461_1462delAG (c.1457_1458delAG) | p.Arg487SerfsTer19 (p.K486fs*20) |
| c.1597_1598dupT (c.1597_1598insT) | p.Cys533LeufsTer5 (p.L533fs*5) |
| c.1615T>G | p.Trp539Gly |
| c.1674-2A>G | -- |
| c.1741G>C | p.Gly581Arg |
Homo sapiens establishment of sister chromatid cohesion N-acetyltransferase 2 (ESCO2), mRNA
Normal gene product Translation of the mRNA results in a protein of 601 amino acids with two different domains, the C-terminal portion with acetyltransferase activity and the N-terminal end, which binds to chromatin. The acetyltransferase domain is homologous to Drosophila deco and S cerevisiae eco1, which are proposed to play a role in establishing sister chromatid cohesion during S phase after DNA replication.
The following graph shows the percentage of amino acids of the protein.

Abnormal gene product The abnormalities reported in ESCO2 are predicted to lead to loss of function, truncation in the protein, or single amino acid changes. Table 2 shows the amino acid changes. The c.1615T>G (pTrp539Gly) missense mutation results in loss of in vitro acetyltransferase activity . The cellular phenotype resulting from this missense mutation is equivalent to the one produced by nonsense and frameshift mutations, indicating that the RBS molecular mechanism involves loss of acetyltransferase activity. Alterations in ESCO2 function result in lack of cohesion at heterochromatic regions, which may lead to activation of the mitotic spindle checkpoint, with the subsequent mitotic delay and impaired cell proliferation observed in RBS cells. The clinical manifestations of RBS may result from the loss of progenitor cells during embryogenesis of structures affected in RBS.
Ctf7p is essential for sister chromatid cohesion and links mitotic chromosome structure to the DNA replication machinery 1999
Yeast cohesin complex requires a conserved protein, Eco1p(Ctf7), to establish cohesion between sister chromatids during DNA replication 1999
Two human orthologues of Eco1/Ctf7 acetyltransferases are both required for proper sister-chromatid cohesion 2005
Two putative acetyltransferases, san and deco, are required for establishing sister chromatid cohesion in Drosophila 2003
Inactivating mutations in ESCO2 cause SC phocomelia and Roberts syndrome: no phenotype-genotype correlation 2005
Roberts syndrome is caused by mutations in ESCO2, a human homolog of yeast ECO1 that is essential for the establishment of sister chromatid cohesion 2005
The molecular mechanism underlying Roberts syndrome involves loss of ESCO2 acetyltransferase activity 2008
TREATMENT
It has been suggested that most individuals with RBS are stillborn or die in infancy. However, it is important to emphasize that, because it is possible for individuals to have normal intelligence and a healthy psychological adjustment, even with all of the stigmata of RBS, such individuals should be managed in a way that allows each to improve their quality of life and to reach their full potential.
Individuals with severe RBS who survive the newborn period face a number of medical problems, and management of these individuals usually requires more than one medical specialist; experts in pediatrics, genetics, ophthalmology, cardiology, nephrology, neurology, child development, rehabilitation, general surgery, orthopedics, or dentistry may be involved.
Treatment is based on the affected individual’s specific needs and may include:
• Surgical treatments including cosmetic or reconstructive surgery for clefts of the lip and/or palate and for limb abnormalities (several surgeries are usually required). Hand surgery improves early and proper development of the prehensile grasp.
• Prostheses
• Speech assessment and therapy and aggressive treatment of otitis media if cleft palate is present
Standard treatment for specific ophthalmologic problems, cardiac defects and renal dysfunction
Surveillance
The following are appropriate:
• Periodic follow up to monitor mental and physical growth and to determine if frequent infections are an issue
• Regular follow up for assessment of speech and ear infections/hearing loss if cleft lip and palate are present
• Annual screening for developmental delays or learning disorders
• Monitoring as per specific ophthalmologic, cardiac, or renal anomalies
Alice Romero