Colorectal cancer (CRC) annually affects 1.000.000 million individuals and causes more than 600.000 deaths. Like most cancers, it is not usually diagnosed until late in life (90% occur after age 55). However routine examination of normal adults with a colonoscopy often reveals a small benign tumor, or adenoma, of the gut epithelium in the form of a protruding mass of tissue, called polyp, that is believed to be the precursor of a large proportion of colorectal cancer. Because the progression of the disease is usually very slow, there is typically a period of about 10 years in which the slowly growing tumor is detectable, but has not yet turned malignant. So, when people are screened by colonoscopy and the polyps are removed, the subsequent incidence of colorectal cancer is very low. If the cancer is not diagnosed, it develops and becomes malignant, cells become invasive and produce metastases. Approximately 20% of the patients have metastatic disease at the time of diagnosis and an additional 20% develop metastases during follow-up.
PATOGENESIS AND PROGRESSION
Colorectal cancer arise from the epithelium lining the colon and rectum. The organization of this tissue is broadly similar to that of small intestine and its renewal depends on stem cells that lie in deep pockets of epithelium, called intestinal crypts. It develops through various stages, some of which are often related to specific gene mutation.
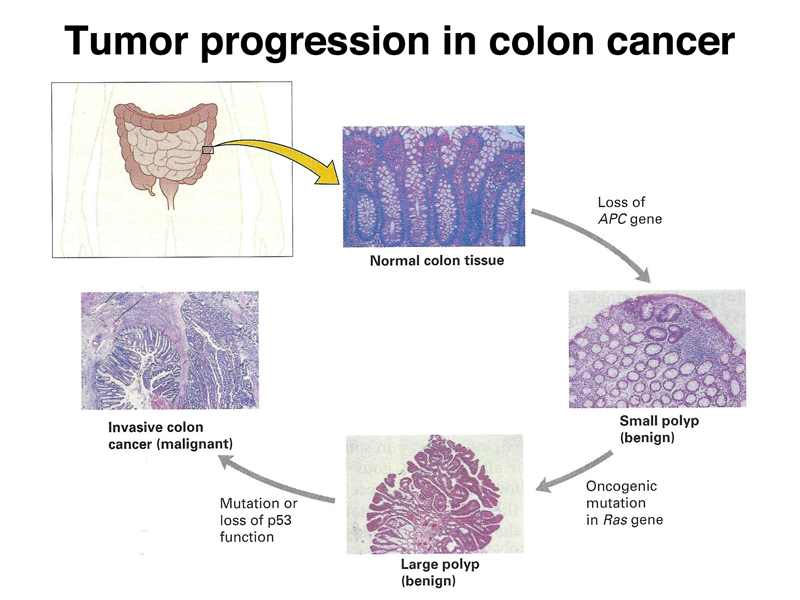
As shown in the image is considered that the loss of function of the APC protein is one of the mutations that determine the tissue hyperplasia that leads to the development of the early adenoma. Additional mutations, particularly those that cause hyperactivation of KRAS protein and inactivation of various tumor suppressor genes (including p53) are responsible for the subsequent progression of the tumor.
Of particular importance to understand the current treatments and issues related to them is KRAS pathway. This pathway is physiologically present in all cells capable of replication (such as stem cells) and is essential for the proliferation and cell survival.
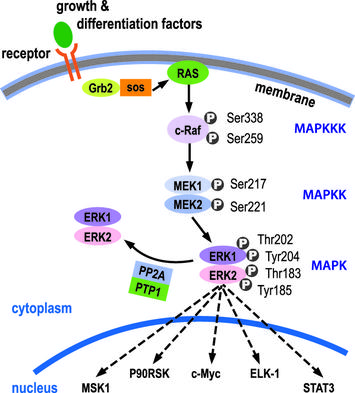
KRAS belongs to the family of RAS proteins, which have GTPase capacity. The activation of KRAS is mediated by growth factors that, by binding to tyrosine kinases receptors on the cell surface, alter their conformation and determine their mutual phosphorylation; this allows the activation of the complex Grb2-SoS that, exchanging the GDP bound to Ras inactive with a molecule of GTP causes the activation. KRAS activates Map-kinases cascade (RAF-MEK-ERK), which activates various targets; the most important is the c-myc that induces progression of the cell in the cell cycle (therefore stimulates the cell proliferation).
If KRAS is mutated (and overactive), the process of proliferation occurs without control, even in the absence of mitogenic stimuli. Generally typical mutations of colorectal cancer are localized in exon 2 (in 90% of cases) or in exons 3 and 4 of the gene (10%) and cause increased functionality of the protein, that result from an overactive domain GTPase or to an increased exchange GDP / GTP. What results is abnormal proliferation that causes the progression of the tumor.
CURRENT THERAPIES AND PREDICTIVE VALUE OF BIOMARKERS
The initial therapy for colorectal cancer, a monotherapy based on a single drug, the 5-fluorouracil, was later replaced by a combined chemotherapy consisting in three drugs (5-fluorouracil, irinotecan and oxaliplatin). The current first-line therapy is represented by the administration of the combined chemotherapeutic, FOLFIRI (5-fluorouracil, folic acid, irinotecan) and a biological agent, a monoclonal antibody (bevacizumab) capable of specifically binding to EGFR (Epidermal growth factor receptor), the receptor of an important growth factor (EGF). Through its receptor, EGF is able to activate the RAS-MAPkinase and PI3K-AKT-mTOR pathways, both capable of stimulating tumor proliferation, invasion, migration, and neovascularization. Thanks to the use of anti-EGFR monoclonal antibodies (such as bevacizumab), it is possible to prevent activation of the receptor by mitogens and so prevent the uncontrolled growth of the tumor.
EGFR blockage elicits multiple downstream effects, primarily modulated by the RAS-MAPK and PI3K-AKT-mTOR signalling pathways. Rational use of targeted therapies requires optimal selection of patients whose tumors are dependent on these pathways.
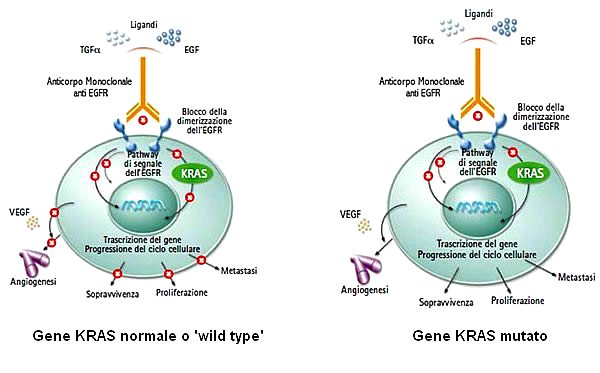
The mutations that were first used to predict the efficacy of treatment are those of KRAS. Wild type individuals (without mutation in the KRAS gene) respond better to the above-mentioned treatment than individuals who have a mutation in the KRAS gene. These latter, in fact, have a reduced response because the mutation causes a constitutive activation of the KRAS protein (and consequently of the downstream pathway), which makes it insensitive to inactivation of the receptor. Already in 2009 some protocols foresaw the need to test the presence of KRAS mutations before deciding the treatment to be followed; in the presence of mutation (35-45% of cases) therapy with FOLFIRI and bevacizumab is strongly discouraged because of limited effectiveness.
Further studies have consequently found a moderate response even in wild type for KRAS individuals, suggesting the existence of other mutations able to modify the outcome of the therapy.
The treatment predictive importance of mutation outside of KRAS exon 2 has gradually recognized and regards NRAS (mutations in codons 12, 13, 61, 146) , BRAF (V600E), PIK3CA and PTEN (mutation or loss); mutations in these markers are found in 4-27% of tumors.
CONCLUSION
The independent predictive values from KRAS, NRAS, BRAF, PIK3CA and PTEN and the increased response rates in tumors that remain wild type after multiple biomarker assessment, strongly suggest treatment predictive testing in CRC should apply biomarker panel in order to optimize identification of patient who will benefit from anti-EGFR treatment.
Testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy
FOLFIRI-bevacizumab as first-line chemiotherapy in patient with advanced colorectal cancer
The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer