INTRODUCTION
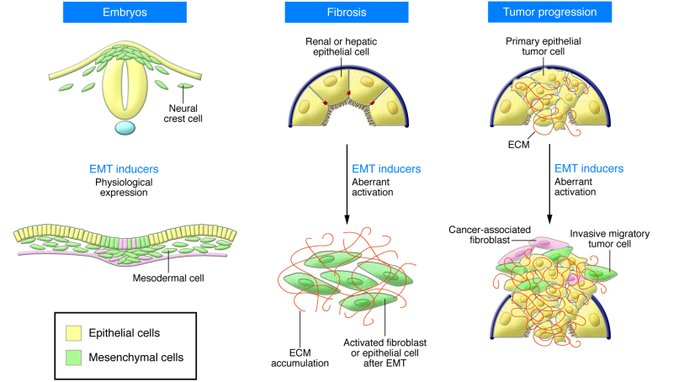
An epithelial-mesenchymal transition (EMT) is a biologic process that allows a polarized epithelial cell, which normally interacts with basement membrane via its basal surface, to undergo multiple biochemical changes that enable it to assume a mesenchymal cell phenotype, which includes enhanced migratory capacity, invasiveness, elevated resistance to apoptosis, and greatly increased
production of ECM components. The completion of an EMT is signaled by the degradation of underlying basement membrane and the formation of a mesenchymal cell that can migrate away from the epithelial layer in which it originated.
EMTs are encountered in three distinct biological settings that carry very different functional consequences. While the specific signals that delineate the EMTs in the three discrete settings are not yet clear, it is now well accepted that functional distinctions are apparent.
The “type 1” neither causes fibrosis nor induces an invasive phenotype resulting in systemic spread via the circulation. Among other outcomes, these type 1 EMTs can generate mesenchymal cells (primary mesenchyme) that have the potential to subsequently undergo a MET to generate secondary epithelia. The EMTs associated with wound healing, tissue regeneration, and organ fibrosis are of a “second type”.
“Type 3” EMTs occur in neoplastic cells that have previously undergone genetic and epigenetic changes, specifically in genes that favor clonal outgrowth and the development of localized
tumors. These changes, notably affecting oncogenes and tumor suppressor genes, conspire with the EMT regulatory circuitry to produce outcomes far different from those observed in the other
two types of EMT. Carcinoma cells undergoing a type 3 EMT may invade and metastasize and thereby generate the final, life-threatening manifestations of cancer progression.
(The basics of epithelial-mesenchymal transition.2009)
Type 1 EMT: embryogenesis
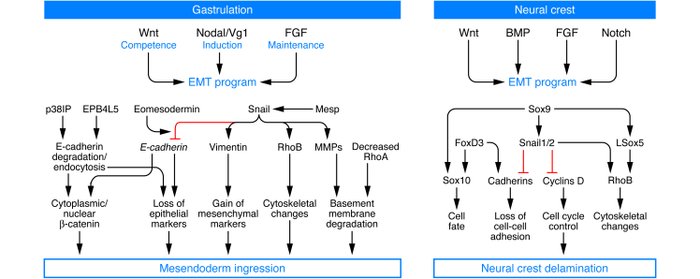
A fertilized egg undergoes gastrulation, generating three germ layers. Once formed, the primitive streak, acting via invagination or ingression, generates the mesendoderm, which subsequently separates to form the mesoderm and the endoderm via an EMT (also known as epiblast-mesoderm
transition) by replacing the hypoblast cells.
Cells of the primary mesenchyme exhibit enhanced migratory properties when compared with those
of the epiblast and the hypoblast.
At the biochemical level, the EMT associated with gastrulation is dependent on and orchestrated by canonical Wnt signaling, and embryos deficient in Wnt3 cannot undergo the EMT associated
with gastrulation. The subsequent formation of the primitive streak is associated with expression of Wnt8c, and ectopic expression of Wnt8c in embryos leads to multiple primitive streaks. TGF-β superfamily proteins, notably Nodal and Vg1, mediate the action of Wnts, and their deficiencies can lead to mesodermal defects due to the absence of functional EMTs. Wnts also cooperate with FGF receptors to help regulate an EMT associated with gastrulation. The Snail, Eomes, and Mesps transcription factors orchestrate the EMT associated with gastrulation.
The signaling pathways utilized during EMT in the neural crest are similar to those that are active at the primitive streak during gastrulation. Indeed, a combination of Wnt, FGF and bone morphogenetic protein (BMP) signaling pathways is necessary for neural crest induction. Experimental evidence has long demonstrated that Notch signaling is also necessary for cranial neural crest development.
(The basics of epithelial-mesenchymal transition.2009)
(Epithelial-mesenchymal transitions: the importance of changing cell statein the development and disease.2009)
Type 2 EMT: organ fibrosis
Such EMTs are found to be associated with fibrosis occurring in kidney, liver, lung, and intestine.
It is found direct evidence for epithelial cells serving, via EMTs, as important precursors of the fibroblasts that arise during the course of organ fibrosis.
Such cells are likely to represent the intermediate stages of EMT, when epithelial markers continue to be expressed but new mesenchymal markers have already been acquired. The behavior of these cells provided one of the first indications that epithelial cells under inflammatory stresses can advance to various extents through an EMT, creating the notion of “partial EMTs”.
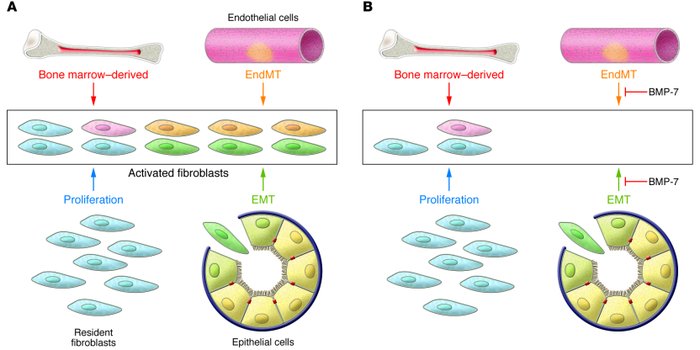
Recent experiments in mice have demonstrated that endothelial cells associated with the microvasculature can also contribute to the formation of mesenchymal cells during the course of fibrosis, doing so via an analogous process known as EndMT. This pathologic process echoes a similar normal process occurring during development.
Lineage-tagging experiments and bone marrow transplant studies demonstrated that during the course of kidney fibrosis in mice, about 12% of fibroblasts are derived from the bone marrow and about 30% are derived via EMT from the tubular epithelial cells of the kidney. In addition, it has been shown more recently that in kidney fibrosis about 35% of fibroblasts are derived via EndMT from the endothelial cells normally residing within the kidney. The remaining portions are speculated to arise via activation of resident fibroblasts or other mesenchymal cells.
Inflammatory injury to the mouse kidney can result in the recruitment of a diverse array of cells that can trigger an EMT through their release of growth factors, such as TGF-β, PDGF, EGF, and FGF-2.
β1 integrin and integrin-linked kinase (ILK) are also identified as important mediators of the TGF-β–induced EMT associated with tubular epithelial cells.
TGF-β induces EMT via both a Smad2/3-dependent pathway and a MAPK-dependent pathway. Recent experiments have also demonstrated a key role for the E-cadherin/β-catenin signaling axis
for EMT involving epithelial cells.
Systemic treatment of mice with renal fibrosis with recombinant human BMP-7 reverses renal disease due to severe attenuation of the formation of EMT- and EndMT-derived fibroblasts.
(The basics of epithelial-mesenchymal transition.2009)
Type 3 EMT: cancer progression and metastasis
Activation of an EMT program has been proposed as the critical mechanism for the acquisition of malignant phenotypes by epithelial cancer cells.
Many mouse studies and cell culture experiments have demonstrated that carcinoma cells can acquire a mesenchymal phenotype and express mesenchymal markers such as α-SMA, FSP1, vimentin and desmin.
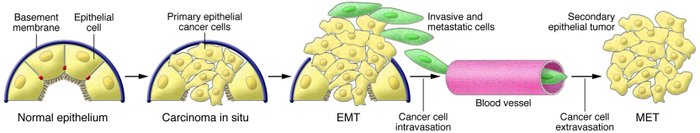
An apparent paradox comes from the observation that the EMT-derived migratory cancer cells typically establish secondary colonies at distant sites that resemble, at the histopathological
level, the primary tumor from which they arose; accordingly, they no longer exhibit the mesenchymal phenotypes ascribed to metastasizing carcinoma cells.
Reconciling this behavior with the proposed role of EMT as a facilitator of metastatic dissemination
requires the additional notion that metastasizing cancer cells must shed their mesenchymal phenotype via a MET during the course of secondary tumor formation.
One suggestion is that the genetic and epigenetic alterations undergone by cancer cells during
the course of primary tumor formation render them especially responsive to EMT-inducing heterotypic signals originating in the tumor-associated stroma.
In the case of many carcinomas, EMT-inducing signals emanating from the tumor-associated stroma, notably HGF, EGF, PDGF and TGF-β, appear to be responsible for the induction or functional activation in cancer cells of a series of EMT-inducing transcription factors, notably Snail, Slug, zinc finger E-box binding homeobox 1 (ZEB1), Twist, Goosecoid, and FOXC2. Once expressed and activated, each of these transcription factors can act pleiotropically to choreograph the complex EMT program.
TGF-β is an important suppressor of epithelial cell proliferation and thus primary tumorigenesis. However, it is now clear that in certain contexts it can also serve as a positive regulator of tumor
progression and metastasis.
Two possible signaling pathways have been identified as mediators of TGF-β–induced EMT. The first of these involves Smad proteins, which mediate TGF-β action to induce EMTs via the ALK-5 receptor. Smad-mediated signaling induced by TGF-β facilitates motility.
A second TGF-β–induced pathway in EMT is also compelling. More specifically, some data indicate that p38 MAPK and RhoA mediate an autocrine TGF-β–induced EMT in NMuMG mouse mammary epithelial cells. This process also requires integrin β1–mediated signaling and
the activation of latent TGF-β by αVβ6 integrin.
The connection between inflammation and EMT was demonstrated when COX-2 was shown to inactivate Smad signaling and enhance EMT stimulated by TGF-β through a PGE2-dependent mechanism. In inflammation, macrophages and activated resident fibroblast release chemokines and MMP2-MMP3-MMP9, that induce basament membrane damage and focal degradation of type IV collagen and laminin.
Epithelial cell adhesion complexes reorganize and cell proliferation is suppressed when the full-length or the cytoplasmic portion of E-cadherin (containing the β-catenin binding site) is ectopically espresse in cells that have passed through an EMT, causing such cells to lose their mesenchymal phenotype. Sequestration of β-catenin in the cytoplasm is important for the preservation of epithelial features of cancer cells, and acquisition of the mesenchymal phenotype correlates with the movement of β-catenin to the nucleus, where it becomes part of Tcf/LEF complexes. Such β-catenin accumulation in the nucleus, which is often associated with loss of E-cadherin expression, correlates with susceptibility to enter into an EMT and acquisition of an invasive phenotype. Thus, cells that lose cell surface E-cadherin become more responsive to induction of an EMT by various growth factors.
The central role played by E-cadherin loss in the EMT program is further illustrated by the actions of several EMT-inducing transcription factors that facilitate acquisition of a mesenchymal phenotype, such as Snail and Slug, as well as those encoding two key zinc finger–containing basic helix-loop-helix transcription factors, serviva of motor neuron protein interacting protein 1 (SIP1) and E12.
Loss of E-cadherin promotes Wnt signaling and is associated with high levels of Snail in the nucleus. SIP1 represses E-cadherin expression and binds, along with Snail, to the E-cadherin promoter in an overlapping fashion.


(The basics of epithelial-mesenchymal transition.2009)
EMT and MicroRNAs
MicroRNAs (miRNAs) are a class of small highly conserved RNAs that provide widespread expressional control through the translational repression of MiRNA. A number of miRNA expression profiling studies have been conducted to identify candidate miRNAs with possible roles in TGFβ-induced EMT.
TGFβ appears to play a dominant role, directly activating transcription factors ZEB, SNAIL
and TWIST, the master regulators of EMT.
During EMT there is a suppression of the miR-200 family of miRNA.

(The switch from E- to N-cadherin is considered typical
for EMT, and enzymatically shed, soluble ectodomains
from both cadherins have been detected at elevated levels
in the serum of cancer patients. Soluble Ncadherin
(sN-cad) may be a very sensitive circulating
marker, because it is upregulated in cancer cells
and in stromal myofibroblasts.)
(Molecular and pathological signatures of epithelial-mesenchymal transitions at the cancer invasion front.2008)
Amongst the most powerful downstream mediators of TGFβ-dependent EMT activation are the
Zinc Finger E-box-binding homeobox (ZEB 1 and 2) transcription factors that act in part by re pressing the transcription of E-cadherin.
MiR-200 directly targets the 3’ UTR of ZEB mRNA, downregulates ZEB expression and promotes epithelial differentiation.
Importantly, however, the common miR-200 family promoter region also contains highly conserved ZEB-binding sites through which ZEB 1 and 2 exert reciprocal control at the level of miR-200 transcription. Based on this evidence, it has been postulated that miR-200 and the ZEB family
of transcription factors are involved in a double negative feedback loop which acts to stabilize
cellular differentiation.

(MicroRNAs: critical regulators of epithelial to mesenchymal (EMT) and mesenchymal to epithelial transition (MET) in cancer progression.2011)
Conclusions
EMT and the reverse process MET are complex, multi-faceted and tightly regulated developmental
programs that play key roles during morphogenesis, but that become usurped during the process of cancer cell metastasis.
Type 3 EMTs occur in the context of tumor growth and cancer progression, when cancer cells at the invasive front of the tumors convert to a mesenchymal phenotype. The induction of type 3 EMTs is facilitated by the genomic alterations acquired by cancer cells and these EMTs generate cells with invasive properties that enable them to move into the blood stream and spread systemically to other organs.
(The basics of epithelial-mesenchymal transition.2009)
(MicroRNAs: critical regulators of epithelial to mesenchymal (EMT) and mesenchymal to epithelial transition (MET) in cancer progression.2011)