DEFINITION
Bradykinin is an inflammatory mediator consisting of nine amino acids.

It is a peptide that causes blood vessels to dilate (enlarge), and therefore causes blood pressure to fall. A class of drugs called ACE inhibitors, which are used to lower blood pressure, increase bradykinin (by inhibiting its degradation) further lowering blood pressure. Bradykinin dilates blood vessels via the release of prostacyclin, nitric oxide, and Endothelium-Derived Hyperpolarizing Factor.
DEFINITION
BRADYKININ is a nine-amino acid peptide with a molecular weight of 1.06 kDa.
It is part of the kinin–kallikrein system and plays different roles by contributing to inflammatory responses, influencing blood with its activities and increasing pain signals.
Its mechanisms are still to be completely understood.
THE GENE
CHEMICAL STRUCTURE AND IMAGES
Primary Structure
It’s a nonapeptide whose sequence consists of these nine aminoacids: R-P-P-G-F-S-P-F-R
Tertiary Structure

Protein Aminoacids Percentage

SYNTHESIS
The gene of the bradykinin does not exist: it is the product of a post-transcriptional edit and post-translational modifications performed by various enzymes.
The first step is the transcription of the KNG1 gene (Kininogen Gene 1)found on chromosome 3q27.
KNG1 interactions
Two different alternative splices are used to generate two different proteins: the High Molecular Weight Kininogen (also known as HMWK) and Low Molecular Weight Kininogen (LMWK).


The former is also called "Williams-Fitzgerald-Flaujeac factor" and it is involved in blood coagulation through the intrinsic way ( High-molecular weight kininogen and the intrinsic coagulation pathway in patients with de novo acute myocardial infarction,2013) as well as in the kinin-kallikrein system ( Plasma kallikrein: the bradykinin-producing enzyme, 2013).
High-molecular weight kininogen and the intrinsic coagulation pathway in patients with de novo acute myocardial infarction,2013) as well as in the kinin-kallikrein system ( Plasma kallikrein: the bradykinin-producing enzyme, 2013).
The latter has not been functionally understood yet but It has been seen to be together with tissue kallikrein so that lys-bradykinin is produced; an aminopeptidase has the role to convert this peptide into bradykinin by getting rid of the N-terminal lysine.
HMWK is produced by the liver as well as prekallikrein.
However human vascular endothelial cells contain the highest fraction of mRNA for HMWK and some kininogen has been found in rat vascular smooth muscle cells ( Kallikreins and Kinins: Some Unanswered Questions About System Characteristics and Roles in Human Disease, 1995)
Kallikreins and Kinins: Some Unanswered Questions About System Characteristics and Roles in Human Disease, 1995)

| A plot showing the tissues in which KNG1 gene (the gene of bradykinin precursors) is expressed |
When the Hageman Factor (or Factor XII) is activated it cleaves prekallikrein to produce kallikrein; then HMWK is to be complexed with kallikrein and Factor XII so that the intrinsic way is performed.
Kallikrein also allows the Bradykinin to be producted by proteolytic cleavage of HMWK.
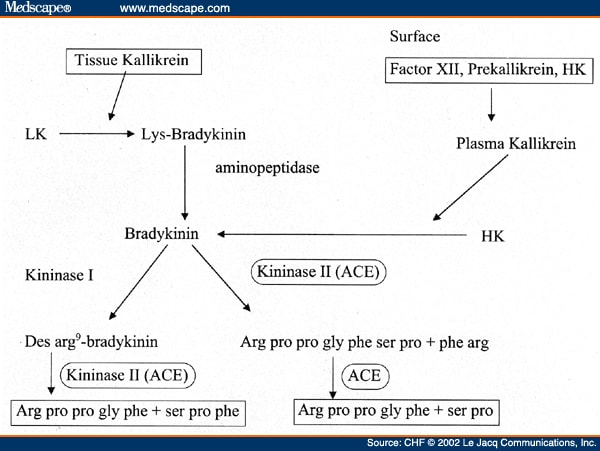
| This image shows the ways in which the bradykinin is both produced and turned over |
TURNOVER
In mammals the bradykinin can be turned over by following two pathways.
The former is also the main one and it is performed by three enzymes: Angiotensin Converting Enzyme (ACE), N-Carboxypeptidase (CPN) and Neutral Endopeptidase (NEP) (  Inactivation of bradykinin by angiotensin-converting enzyme and by carboxypeptidase N in human plasma,2000).
Inactivation of bradykinin by angiotensin-converting enzyme and by carboxypeptidase N in human plasma,2000).

| An image showing the sites of cleavage performed by enzymes |
ACE is the most important enzyme for the inactivation of Bradykinin due to the turn-over plot.
In fact, once produced bradykinin rapidly decreases its blood concentration while a big rise of 1-5 bradykinin peptide performed by ACE is seen.
As shown below, ACE activity is performed twice as it firstly cleaves the bradykinin so that a 1-7 bradykinin is produced; as soon as this process is completed it performes its last activity by cutting this peptide and giving the 1-5 bradykinin.

The plots show how the inactivation of bradykinin is carried out by the angiotensin-converting-enzyme for almost 65 per cent of the whole activity while the other two enzymes (CPN and NEP) turn bradykinin over for 32 per cent and three per cent respectively.
The latter is a way whose enzyme is the Kininase I: it turns the bradykinin into des-arg 9- bradykinin which is then turned over by ACE
( Bradykinin and des-Arg9-bradykinin metabolic pathways and kinetics of activation of human plasma,2001)
BIOLOGICAL FUNCTIONS
The bradykinin is a nonapeptide; yet it cohoperates with such a huge amount of pathways they are still to be completely understood.
In the kinin system there are two receptors (GPCRs) whose principal ligand is the protein bradykinin; they are known as BR1, or B1 and BR2, or B2 (Bradykinin Receptor 1 and 2, respectively).
While B2 is constitutively expressed in many tissues, B1 is thought to be induced under inflammatory conditions; however B1 and B2 receptors are always expressed in the hippocampus of humans and seem to be overexprssed in people with temporal lobe epilepsy
( B2 receptor analysis, 2014).
B2 receptor analysis, 2014).
The Bradykinin Receptor 2 plays an important role in multiple pathways through bradykinin ability to elevate vascular permeability and to cause vasodilatation in blood vessels.


The bradykinin allows the vessel to increase its permeability through the interactions with B2: since it is a receptor coupled with a G protein the ligand let the activity begins. In the citosolic side of the cell Gi activates PLC which allows PIP2 to turn into IP3 and DAG. The former makes the calcium to increase so that CaM kinase is activated while DAG lets PKC to be functionally operative as well.
These kinases activate the PLA2 which performes the synthesis of the arachidonic acid; throughout this way PGI2 is produced and the first bradykinin effect in vessels dilatation.
CaM K also activates eNOS whose substrate is L-Arg; it makes both Nitric Oxide (NO) and citrulline. Nitric Oxide allows the Guanylate Cyclase to produce cGMP from GTP. cGMP activates the PKG which phosphorilates PD III (Phosphodiesterase III).
This protein allows cAMP to become AMP (everywhere but air ways smooth muscle cells) so that PKA is active no more and L Channels cannot allow any Ca2+ flow from the outer side of the cell. As result of this process the smooth muscle cells of the blood vessels have their walls swelled.
As the image above shows, the bradykinin has an important role in pro-inflammatory response.
Recent studies demonstrate the bradykinin induces IL-6 expression in airway smooth muscles cells by involving the activation of ERK.
( The kinin system - bradykinin: biological effects and clinical implications. Multiple role of the kinin system - bradykinin,2007)
Nontheless this peptide is involved in cAMP synthesis via MAPK regulation of cytosolic phospholipase A2 and prostaglandin E2 release in airway smooth muscle.
( Bradykinin stimulates cAMP synthesis via mitogen-activated protein kinase-dependent regulation of cytosolic phospholipase A2 and prostaglandin E2 release in airway smooth muscle,1997)
As a result of this we can see both an inflammatory response and an increase in vessels permeability since a disassembly of adherens and tight-junction is performed. In fact not only does the bradykinin involves the IL-6 pathway but it also activates the NF-kB way in order to promote vessel growth and remodeling. NFkB leades to a massive COX-2 overexpression so that VEGF is functionally operative.
( Antagonism of bradykinin B2 receptor prevents inflammatory responses in human endothelial cells by quenching the NF-kB pathway activation, 2014)
These elements have the adherens and tight-junction disassembled in a certain percentage so that the increased permeability makes the vessel less opposing to the pressure found in it.
This is the very beginning of edema process.
In bronchi these leaps allow a quantity of fluid to gather into the intersitium. Nonetheless the bradykinin causes bronchoconstriction
through its B2 receptor ( Inhibition of bradykinin-induced bronchoconstriction in the guinea-pig by a synthetic B2 receptor antagonist,1989).
However not only does the BK cause the bronchial edema but it can also allow people to develop angioedema.
Since ACE-Inhibitors are often used for hypertension and its treatment such side effects are seen in one patient out of five.

| Angioedema induced by treatment with ACE-inhibitors |
In respiratory system such effects brings to dry cough: the fluid gathered in interstitium stimulates the cough reflex in about one patient out of ten until the patient himself stops taking this drug. As a result of this side effect sartans are usually preferred.
ACE-INHIBITORS INDUCED COUGH
The origin of the cough is to be related to vagus-innerved sites in which there are cough receptors as well.
These receptors can be divided into four groups:
TRPV1 (transient receptor potential vanilloid)
TRPA1 (transient receptor potential ankyrine),
RAR (rapid adapting receptors)
SAR (slow adapting receptors).

| A simple image showing the main elements of cough generating pathways |
TRPV1 receptors can be stimulated by both capsaicin and citric acid (or in general by an increase of [H+]); there are evidences of the cold induced activation of TRPA1 whose main function is to increase the sensibility of such areas.
While the previous receptors can be considered as minor, RAR is to be thought as the main one. It is linked to myelin-sheathed fibres and can be only found in larynx.
The last receptors we find are the SAR. They are set into the smooth muscle cell in the respiratory system and linked to the same kind of fibres of RARs.
Despite having described the functionally main receptors there is another kind of fibres which is numerically more seen: C-fibres; they are not myelin sheathed yet they are the 80% of the total amount in this area.
A common element is seen: all these elements have the same final destination: the nucleus of the solitary tract (NTS) which is the brainstem. As of today it is considered the “cough generator” since it stimulates all the muscles involved into the respiration through both the phrenic nerve and the recurrent laryngeal nerve.
In such a scenario it is possible to understand the reason ACE-I induced cough can exist for: as written above BK is no longer properly turned over in ACE-I therapy and co-operates to pro-inflammatory response as well as it causes oedema and fluid gathering throughout the respiratory epithelium. PGE2 (produced by COX2 BK-induced overexpression) and all the others inflammatory elements stimulates the explained receptors so that NTS neurons receive a pro-coughing response and begins the stimulation of the proper muscles.
As a result of these processes there is no overproduction of mucus made by goblet cells so that we can see a dry cough in a certain percentage of patients.
( Prevalence, pathogenesis, and causes of chronic cough,2008)
THERAPEUTIC USE
Despite being a strong anti-hypertensive molecule the Bradykinin has not been approved for any kind of therapeutic use since its side effects are too widely and too hard to control.
Althoug its inhibitor, the Icatibant was granted FDA approval on August 25th, 2011 fot injection use in treatment of Hereditary Angioedema
(25 Aug 2011 FDA Approves Shire’s FIRAZYR® icatibant injection for Acute Attacks of Hereditary Angioedema HAE)

| This picture illustrates the effects of icatibant-induced selective block of B2 receptors in treatment of Angioedema after its injection and 1 to 4 hours checkpoints |
ACE Inhibitor-Induced Angioedema, 1997)

Bi-directional actions of estrogen on the renin-angiotensin system 1999
- Estrogen stimulates the renin-angiotensin system by augmenting both tissue and circulating levels of angiotensinogen and renin. We show, however, that angiotensin converting enzyme (ACE) activity in the circulation and in tissues is reduced in two animal models of postmenopausal chronic hormone replacement. We observed a reduction of ACE activity in association with a significant increase in plasma angiotensin I (Ang I) and hyperreninemia in ovariectomized monkeys treated with Premarin (conjugated equine estrogen) replacement for 30 months. Plasma angiotensin II (Ang II) levels were not increased in monkeys treated with estrogen, suggesting that the decrease in ACE curtailed the formation of the peptide. The Ang II/Ang I ratio, an in vivo index of ACE activity, was significantly reduced by estrogen treatment, further supporting the biochemical significance of estrogen's inhibition of ACE. In ovariectomized transgenic hypertensive (mRen2)27 rats submitted to estrogen replacement treatment for 3 weeks, ACE activity in plasma and tissue (aorta and kidney) and circulating Ang II levels were reduced, whereas circulating levels of angiotensin-(1-7) (Ang-(1-7) were increased. Ang-(1-7), the N-terminal fragment of Ang II, is a novel vasodilator and antihypertensive peptide. Thus, the net balance of these effects of estrogen on the renin-angiotensin vasoconstrictor/vasodilator system is to promote the antihypertensive effect.
Mechanisms of cyclosporine A toxicity