The androgen receptor (AR) is a type of nuclear receptor (NR) which is activated by binding of either the androgenic hormones testosterone or dihydrotestosterone in the cytoplasm and then translocating into the nucleus. The NRs are a class of structurally related proteins that modulate gene expression by acting as ligand-dependant transcription factors. NR ligands exert their effects by binding to an intracellular steroid hormone receptor. As for certain NR ligands the action is occurring in a tissue selective manner, the term “selective receptor modulator” has been given to these molecules, since the term “modulators” refers to compounds that have a spectrum of activities ranging from full agonism to partial agonism to full antagonism.
Steroidal NR ligands are known to play important roles in the health of both men and women. In regard to men’s health, testosterone (T, fig. 1) and dihydrotestosterone (DHT) are endogenous steroidal ligands for the AR that likely play a role in every tissue type found in the mammalian body. During the development of the fetus, androgens play a role in sexual differentiation and development of male sexual organs. Furthermore, cognitive function, sexuality, aggression and mood are some of the behavioral aspects mediated by androgens. The main metabolic pathways for T are shown in fig. 2.

Fig. 1 Testosterone

Fig. 2 Metabolic pathways for Testosterone
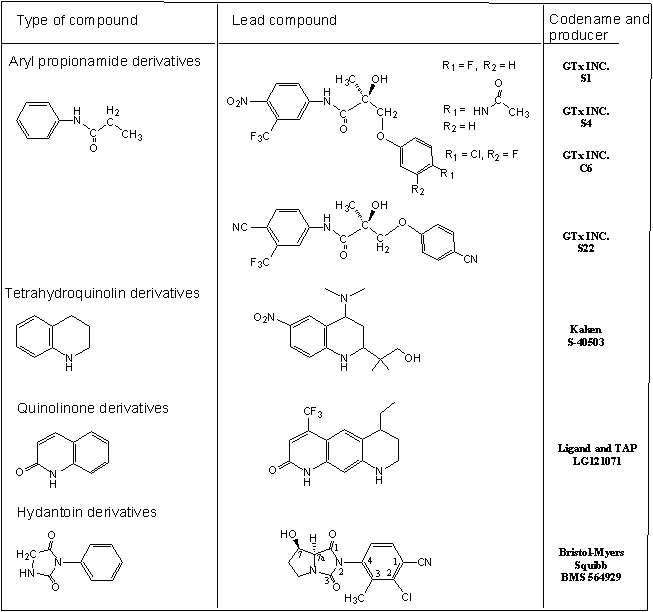
The study of androgen action and male reproductive dysfunction continues to expand significantly. Clinical manifestations include fatigue, depression, decreased libido, erectile dysfunction as well as changes in cognition and mood. Published information indicates that androgen replacement therapy (ART) in men may have benefits in terms of improving body composition parameters as well as improving libido and mood in some men. Current testosterone-based ARTs suffer from rather severe limitations. When dosed orally, unmodified T is highly absorbed, but quickly cleared by the liver. Therefore, it is not possible to achieve the exposures required to see pharmacological effects. The two major strategies to circumvent the metabolic degradation of testosterone are the parenteral administration of C17-OH testosterone esters, and the preparation of 17-alpha alkylated derivatives. Esterified T preparations, such as T propionate, act as a slow release depots of T when given as injections. In general, current ARTs fail to correctly mimic physiological testosterone levels and have potential side effects. Furthermore, ever since the importance of serum testosterone levels for muscle strength and physical performance became evident, the development of tissue-selective and orally bioactive anabolic agents has been a pharmaceutical challenge. To counteract the age-related functional decline in men, not only symptoms of frailty but also muscle wasting, numerous derivative of anabolic androgenic steroids were synthesized and clinically tested. Although beneficial effects such as improved lean body mass and muscle strength were observed, clinical concerns about potential side effects including benign prostatic hypertrophy and occult prostate cancer limited the utility of these substances. Hence, numerous research groups focused on the development of nonsteroidal, tissue-selective anabolic agents with significantly reduced androgenic side effects. Compounds with Selective Androgen Receptor Modulators (SARMs) are currently categorized into ayil propionamides, tricyclic quinolines, tetrahydroquinolines and bicyclic hydantoines (fig. 3).
Major advantages of these drug candidates are enormous tissue-selective anabolic properties combined with considerably reduced side effects commonly associated with steroid replacement therapies such as gynecomastia, decreased levels of HDL cholesterol and hepatic toxicity. This is, at least in part, because these drugs do not undergo the metabolic reactions of testosterone and analogous compounds, meaning that they are not aromatizable or substrates for 5α-reductase. Recent receptor-binding studies have provided additional insights into potential mechanism underlying the tissue selectivity of SARMs. Ligand binding to the AR was shown to induce conformational changes in the ligand-binding domain corresponding to the particular structure of the drug. As a consequence, the altered surface topology apparently enables interactions with different cofactors, which contribute to a tissue-specific gene regulation. Tissue specificity contributes to the enormous medical utility of SARMs, but also increase the temptation to misuse these highly effective anabolic agents to artificially enhance physical performance.
The potential of SARMs for being misused in sport is considered very high in particular, since anabolic agents have represented the most frequently detected class of drugs misused in sports for more than two decades. Hence, doping control laboratories are obliged to implement detection assays capable of detecting most advanced representatives of these new therapeutics. Until now, no SARM is commercially available but illicit market, as few have passed phase-II clinical trials.
Dalton JT, Biochem. Biophys. Res. Commun., 1998, 244, 1-4
Cadilla R. & Turnbull P., Curr. topics in Med. Chem., 2006, 6, 245-270
Evans RM, Science, 1988, 240, 889-895
Thevis M. Et al., Drug discov. today, 2008, 13, 59-66
Smith CL & O’Malley BW, Endoc. Rev., 2004, 25, 45-71
Mohler ML et al., Exp. Opin. Ther. Patents, 2005, 15, 1565-1585
Gao W & Dalton JT, Mol. Interv., 2007, 7 10-13
Thevis et al., J. Mass Spec., 2009, 44, 442-460