11β-Hydroxysteroid dehydrogenase (HSD-11β or 11β-HSD) is the name of a family of enzymes that catalyze the conversion of inert 11 keto-products (cortisone) to active cortisol, or vice versa, thus regulating the access of glucocorticoids to the steroid receptors.
11β-hydroxysteroid + NADP+ an 11-oxosteroid + NADPH + H+
Thus, the two substrates of this enzyme are 11beta-hydroxysteroid and NADP+, whereas its 3 products are 11-oxosteroid, NADPH, and H+.
This enzyme belongs to the family of oxidoreductases, specifically those acting on the CH-OH group of donor with NAD+ or NADP+ as acceptor. The systematic name of this enzyme class is 11beta-hydroxysteroid:NADP+ 11-oxidoreductase. Other names in common use include corticosteroid 11beta-dehydrogenase, beta-hydroxysteroid dehydrogenase, 11beta-hydroxy steroid dehydrogenase, corticosteroid 11-reductase, and dehydrogenase, 11beta-hydroxy steroid. This enzyme participates in c21-steroid hormone metabolism and androgen and estrogen metabolism.
Function
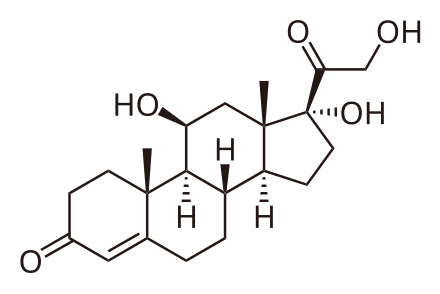 Cortisol. Note the OH at the 11 position on ring C.
Cortisol. Note the OH at the 11 position on ring C.
 Cortisone
Cortisone
Cortisol, a glucocorticoid, binds the glucocorticoid receptor. However, because of its molecular similarity to aldosterone it is also capable of binding the mineralcorticoid receptor. Both aldosterone and cortisol have a similar affinity for the mineralocorticoid receptor; however, there is vastly more cortisol in circulation than aldosterone. To prevent over-stimulation of the mineralocorticoid receptor by cortisol, HSD-11β converts the biologically active cortisol to the inactive cortisone, which can no longer bind to the mineralocorticoid receptor. HSD-11β co-localizes with intracellular adrenal steroid receptors. Licorice or Carbenoxolone , which contains glycyrrhetinic acid, can inhibit 11β-HSD and lead to a mineralocorticoid excess syndrome.
11beta-hydroxysteroid dehydrogenase and its role in the syndrome of apparent mineralocorticoid excess.
Source(2001)
Department of Pediatrics, University of Texas Southwestern Medical Center, 75390-9063, USA. perrin.white@utsouthwestern.edu
Abstract
Aldosterone, the most important mineralocorticoid, regulates electrolyte excretion and intravascular volume mainly through its effects on renal cortical collecting ducts, where it acts to increase sodium resorption from and potassium excretion into the urine. Excess secretion of aldosterone or other mineralocorticoids, or abnormal sensitivity to mineralocorticoids, may result in hypokalemia, suppressed plasma renin activity, and hypertension. The syndrome of apparent mineralocorticoid excess (AME) is an inherited form of hypertension in which 11beta-hydroxysteroid dehydrogenase (11-HSD) is defective. This enzyme converts cortisol to its inactive metabolite, cortisone. Because mineralocorticoid receptors themselves have similar affinities for cortisol and aldosterone, it is hypothesized that the deficiency allows these receptors to be occupied by cortisol, which normally circulates at levels far higher than those of aldosterone. We cloned cDNA and genes encoding two isozymes of 11-HSD. The liver or 11-HSD1 isozyme has relatively low affinity for steroids, is expressed at high levels in the liver but poorly in the kidney, and is not defective in AME. The kidney or 11-HSD2 isozyme has high steroid affinity and is expressed at high levels in the kidney and placenta. Mutations in the gene for the latter isozyme have been detected in all kindreds with AME. Moreover, the in vitro enzymatic activity conferred by each mutation is strongly correlated with the ratio of cortisone to cortisol metabolites in the urine, with age of diagnosis, and with birth weight. This suggests that the biochemical and clinical phenotype of AME is largely determined by genotype.
Glycyrrhizic acid suppresses type 2 11 beta-hydroxysteroid dehydrogenase expression in vivo.
Source(2002)
Third Department of Internal Medicine, Gifu University School of Medicine, 40 Tsukasa-machi, Gifu 500-8705, Japan.
Abstract
Licorice-derivatives such as glycyrrhizic acid (GA) competitively inhibit 11 beta-hydroxysteroid dehydrogenase(11 beta-HSD) type 2 (11-HSD2) enzymatic activity, and chronic clinical use often results in pseudoaldosteronism. Since the effect of GA on 11-HSD2 expression remains unknown, we undertook in vivo and in vitro studies. Male Wistar rats were given 30, 60 or 120 mg/kg of GA twice a day for 2 weeks. Plasma corticosterone was decreased in those given the 120 mg dose, while urinary corticosterone excretion was increased in those given the 30 and 60 mg doses but decreased in those given 120 mg GA. NAD-dependent dehydrogenase activity in kidney microsomal fraction was decreased in animals receiving doses of 60 and 120 mg GA. The 11-HSD2 protein and mRNA levels were decreased in those given 120 mg GA.
 In contrast, in vitro studies using mouse kidney M1 cells revealed that 24h treatment with glycyrrhetinic acid did not affect the 11-HSD2 mRNA expression levels. Thus, in addition to its role as a competitive inhibitor of 11-HSD2, the chronic high dose of GA suppresses mRNA and protein expression of 11-HSD2 possibly via indirect mechanisms. These effects may explain the prolonged symptoms after cessation of GA administration in some pseudoaldosteronism patients.
In contrast, in vitro studies using mouse kidney M1 cells revealed that 24h treatment with glycyrrhetinic acid did not affect the 11-HSD2 mRNA expression levels. Thus, in addition to its role as a competitive inhibitor of 11-HSD2, the chronic high dose of GA suppresses mRNA and protein expression of 11-HSD2 possibly via indirect mechanisms. These effects may explain the prolonged symptoms after cessation of GA administration in some pseudoaldosteronism patients.
11β-Hydroxysteroid dehydrogenase type 1, but not type 2, deficiency worsens acute inflammation and experimental arthritis in mice.
Source(2012)
Centre for Cardiovascular Sciences, Queen's Medical Research Institute, University of Edinburgh, 47 Little France Crescent, Edinburgh EH16 4TJ, United Kingdom.
Abstract
Glucocorticoids profoundly influence immune responses, and synthetic glucocorticoids are widely used clinically for their potent antiinflammatory effects.
 Endogenous glucocorticoid action is modulated by the two isozymes of 11β-hydroxysteroid dehydrogenase (11β-HSD). In vivo, 11β-HSD1 catalyzes the reduction of inactive cortisone or 11-dehydrocorticosterone into active cortisol or corticosterone, respectively, thereby increasing intracellular glucocorticoid levels. 11β-HSD2 catalyzes the reverse reaction, inactivating intracellular glucocorticoids. Both enzymes have been postulated to modulate inflammatory responses. In the K/BxN serum transfer model of arthritis, 11β-HSD1-deficient mice showed earlier onset and slower resolution of inflammation than wild-type controls, with greater exostoses in periarticular bone and, uniquely, ganglion cysts, consistent with greater inflammation. In contrast, K/BxN serum arthritis was unaffected by 11β-HSD2 deficiency. In a distinct model of inflammation, thioglycollate-induced sterile peritonitis, 11β-HSD1-deficient mice had more inflammatory cells in the peritoneum, but again 11β-HSD2-deficient mice did not differ from controls. Additionally, compared with control mice, 11β-HSD1-deficient mice showed greater numbers of inflammatory cells in pleural lavages in carrageenan-induced pleurisy with lung pathology consistent with slower resolution. These data suggest that 11β-HSD1 limits acute inflammation. In contrast, 11β-HSD2 plays no role in acute inflammatory responses in mice. Regulation of local 11β-HSD1 expression and/or delivery of substrate may afford a novel approach for antiinflammatory therapy.
Endogenous glucocorticoid action is modulated by the two isozymes of 11β-hydroxysteroid dehydrogenase (11β-HSD). In vivo, 11β-HSD1 catalyzes the reduction of inactive cortisone or 11-dehydrocorticosterone into active cortisol or corticosterone, respectively, thereby increasing intracellular glucocorticoid levels. 11β-HSD2 catalyzes the reverse reaction, inactivating intracellular glucocorticoids. Both enzymes have been postulated to modulate inflammatory responses. In the K/BxN serum transfer model of arthritis, 11β-HSD1-deficient mice showed earlier onset and slower resolution of inflammation than wild-type controls, with greater exostoses in periarticular bone and, uniquely, ganglion cysts, consistent with greater inflammation. In contrast, K/BxN serum arthritis was unaffected by 11β-HSD2 deficiency. In a distinct model of inflammation, thioglycollate-induced sterile peritonitis, 11β-HSD1-deficient mice had more inflammatory cells in the peritoneum, but again 11β-HSD2-deficient mice did not differ from controls. Additionally, compared with control mice, 11β-HSD1-deficient mice showed greater numbers of inflammatory cells in pleural lavages in carrageenan-induced pleurisy with lung pathology consistent with slower resolution. These data suggest that 11β-HSD1 limits acute inflammation. In contrast, 11β-HSD2 plays no role in acute inflammatory responses in mice. Regulation of local 11β-HSD1 expression and/or delivery of substrate may afford a novel approach for antiinflammatory therapy.
Ongoing inflammation in Hsd11b1−/− mice 48 h after carrageenan injection. This is a representative images of H&E-stained sections of lungs from Hsd11b1−/− and control mice 48 h after intrapleural injection of carrageenan (100 μl of 0.1% carrageenan) showing, more pronounced lymphoid aggregates (A), persistent thickening of visceral pleura (B), and more aggressive adhesion of lung lobes © in Hsd11b1−/− than in control mice. Images captured at ×50 (A and B) and ×500 © magnification. LA, Lymphoid aggregates; VP, visceral pleura; A, adhesions.
11β-Hydroxysteroid dehydrogenase type 1: relevance of its modulation in the pathophysiology of obesity, the metabolic syndrome and type 2 diabetes mellitus.
Source(2012)
Department of Biochemistry (U38/FCT), Faculty of Medicine, University of Porto, Porto, Portugal.
Abstract
Recent evidence strongly argues for a pathogenic role of glucocorticoids and 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) in obesity and the metabolic syndrome, a cluster of risk factors for atherosclerotic cardiovascular disease and type 2 diabetes mellitus (T2DM) that includes insulin resistance (IR), dyslipidaemia, hypertension and visceral obesity. This has been partially prompted not only by the striking clinical resemblances between the metabolic syndrome and Cushing's syndrome (a state characterized by hypercortisolism that associates with metabolic syndrome components) but also from monogenic rodent models for the metabolic syndrome (e.g. the leptin-deficient ob/ob mouse or the leptin-resistant Zucker rat) that display overall increased secretion of glucocorticoids. However, systemic circulating glucocorticoids are not elevated in obese patients and/or patients with metabolic syndrome. The study of the role of 11β-HSD system shed light on this conundrum, showing that local glucocorticoids are finely regulated in a tissue-specific manner at the pre-receptor level. The system comprises two microsomal enzymes that either activate cortisone to cortisol (11β-HSD1) or inactivate cortisol to cortisone (11β-HSD2). Transgenic rodent models, knockout (KO) for HSD11B1 or with HSD11B1 or HSD11B2 overexpression, specifically targeted to the liver or adipose tissue, have been developed and helped unravel the currently undisputable role of the enzymes in metabolic syndrome pathophysiology, in each of its isolated components and in their prevention. In the transgenic HSD11B1 overexpressing models, different features of the metabolic syndrome and obesity are replicated. HSD11B1 gene deficiency or HSD11B2 gene overexpression associates with improvements in the metabolic profile. In face of these demonstrations, research efforts are now being turned both into the inhibition of 11β-HSD1 as a possible pharmacological target and into the role of dietary habits on the establishment or the prevention of the metabolic syndrome, obesity and T2DM through 11β-HSD1 modulation. We intend to review and discuss 11β-HSD1 and obesity, the metabolic syndrome and T2DM and to highlight the potential of its inhibition for therapeutic or prophylactic approaches in those metabolic diseases.