Written by Giulia Libero and Claudia Pizzo
DEFINITION AND EPIDEMIOLOGY
The neurofibromatoses are genetic disorders that are comprehended in the family of Phakomatoses (or "neurocutaneous syndromes") which are disorders of central nervous system that additionally result in lesions on the skin and the eye. These tissues have a common ectodermal origin.
 Neurofibromatosis
Neurofibromatosis
The neurofibromatoses primarily affect the development and growth of neural (nerve) cell tissues. These disorders cause tumors to grow on nerves and produce other abnormalities such as skin changes and bone deformities. The disorder affects all neural crest cells (Schwann cells, melanocytes and endoneurial fibroblasts).
Neurofibromatosis is an autosomal dominant disorder, which means only one copy of the affected gene is needed for the disorder to develop. Therefore, if only one parent has neurofibromatosis, his or her children have a 50% chance of developing the condition as well. The severity in affected individuals can vary, this may be due to variable expressivity.
Although many inherit the disorder, 30 to 50% of new cases occur spontaneously through mutation (change) in an individual's genes. It affects males and females equally.
Neurofibromatosis was traditionally divided into two entities, central NF and peripheral NF, until it was established in the 1980´s that these entities represent two entirely different clinical disorders due to mutations in two different genes. The National Institute of Health (NIH) Consensus Development Conference held in1987 in Bethesda, Md, USA (NIH 1988) suggested, on the basis of clinical features, two distinct types to be differentiated: NF1 and NF2, and defined the diagnostic criteria for both of them. NF1 was synonymous to the classical von Recklinghausen´s disease, while NF2 corresponded to the predominantly intracranial (central) type of neurofibromatosis.
In the last two decades our knowledge of the natural history, genetics and management of the different forms of neurofibromatosis has changed. Of the numerical classifications of neurofibromatosis proposed in the past, only neurofibromatosis type 1 (Nf1) and neurofibromatosis type 2 (Nf2) have been shown to be distinct at clinical and molecular levels. Mosaicism has been demonstrated both in patients with Nf1 and in patients with Nf2, and features of segmental or mosaic Nf1 and Nf2 have been defined. The outlying phenotypes and the molecular genetics of other, rarer, types of neurofibromatosis have been delineated.
The definition of the different forms of neurofibromatosis depends on the occurrence, number and distribution of CAL spots, tumours of the nervous system (neurofibromas and shwannomas) and ophthalmologic findings (which are frequently asyntomatic). Riccardi in 1982 proposed a classification of neurofibromatosis which included seven different types and an eighth category for cases “not otherwise specified”. This classification has not come into generale use.
Genes differentially expressed in Neurofibromatosis type 1 benign and malignant peripheral nerve sheath tumours
Gorlin et al. Later added two further categories of type VIII “gastrointestinal” and type IX “neurofibromatosis/Noonan” forms.
Viskochil and Carey proposed in 1992 an alternative classification which laid the basis of differentiation by combining clinical and molecular knowledge. They divided the different forms of neurofibromatosis into two broad categories:
- alternate forms having some of the respective clinical features of either Nf1 or Nf2 yet not demostrating the “typical” presentation;
- related forms having classic clinical features of neurofibromatosis in addition to distinctive clinical features not typically seen in either Nf1 or Nf2.
Nf1 tumor suppressor in skin: expression in response to tissue trauma and in cellular differentiation, 2002
At present, however, the most widely used classification continues to be that recommanded in 1987 by the NIH Consensus Conference on Neurofibromatosis. This is a numerical rather than descriptive or eponymic nomenclature, with Nf1 replacing the terms von Recklinghausen, peripheral neurofibromatosis or multiple neurofibromatosis, and Nf2 replacing bilateral acoustic or central neurofibromatosis. The consensus statement acknowledges that are other types which, at that time were not defined well enough to be part of the formal classification. Apart form mosaic/ segmental Nf1 (formerly type V of Riccardi's classification) they are all extremely rare.
The different forms of neurofibromatosis, 1999
NF1 is the more common type of the neurofibromatoses which incidence is about 1/2500-3000. In diagnosing NF1.
NF2 is less common (incidence is about 1/40000). NF2 is characterized by bilateral (occurring on both sides of the body) tumors on the eighth cranial nerve. The tumors cause pressure damage to neighboring nerves. To determine whether an individual has NF2, a physician looks for bilateral eighth nerve tumors and similar signs and symptoms in a parent, sibling, or child.
In most cases, symptoms of NF1 are mild, and patients live normal and productive lives. In some cases, however, NF1 can be severely debilitating. In some cases of NF2, the damage to nearby vital structures, such as other cranial nerves and the brainstem, can be life-threatening.
SYMPTOMS
The clinical manifestations of neurofibromatosis 1 (NF1) are extremely variable.

Early symptoms of Type 1 neurofibromatosis include multiple skin lesions (called cafe au lait spots) that appear during infancy. These lesions can occur anywhere on the body and become more numerous during childhood. Cafe au lait spots also occur in healthy infants and about 10% of the general population has one or two.

During adolescence, benign tumors may develop on the skin (cutaneous), under the skin (subcutaneous), and in connective nerve tissue (neurofibromas). These tumors may be painful and in about 2–5% of cases, they become malignant. Some cutaneous lesions can be pressed into the skin manually (called buttonholing).
Other symptoms of Type 1 NF include the following:
- Curvature of the spine (scoliosis)
- Dizziness
- Enlargement and deformity of the bones (may cause chronic pain)
- Hearing loss
- Learning disabilities
- Tumors of the optic nerve, or optic gliomas (may cause blurry vision, vision loss, and Lisch nodules on the iris)
G. Bartolozzi. NEUROFIBROMATOSI TIPO I, 2009 Medico e Bambino pagine elettroniche 2009
Café-au-lait spots
CAL spots may be present at birth or develop within the first1-2 years of life. Their appearance is often the first sign of the NF1. They increase in number in early childhood, but tend to fade with age or become obscured by numerous dermal neurofibromas. Hence, in adulthood it may be difficult to recognise or count a number greater than six. Neither their number nor their size are related to the severity of the disease.

Café-au-lait (CAL) spots are macules varying in diameter from 0.5 to 50 cm, having typically a smooth contour or, when large, irregular outlines. Their colour varies with background skin pigmentation. CAL spots are not found on the scalp, eyebrows, palms and soles.
They are not unique to Nf1patients and 11%-25% of individuals in the general population have one or two such skin lesions. Clinically, there are no differences between CAL spots in Nf1 patients and those in the general population; it is the increased number that is significant. Few other conditions, however, give rise to more than six typical CAL spots, and are all extremely rare; they include ring chromosome syndrome and Schimke osseous dysplasia.
Skinfold freckling
These are hyperpigmented macules resembling CAL spots that are 1-3 mm in diameter, not related to sun exposure, and seen in the axillae, groins (Crowe’s sign) and around the base of the neck. They can also occur in the inframammary regions and over the trunk. They can also occur in the inframammary regions and over the trunk. They are unique to Nf1, appearing after CAL spots and usually before dermal neurofibromas develop.
They occur most often at 3-5 years of age.
Peripheral neurofibromas
Nearly all adult patients have dermal neurofibromas. Neurofibromas are Schwann cell tumors arising from the fibrous tissue surrounding the peripheral nerve sheath, they are composed of Schwann cells, which are the origin of the tumor.These lie within the dermis and epidermis and move passively with the skin. Most are discrete nodules that are soft, almost gelatinous in consistency, and violaceous in colour. In older patients they tend to increase in size and become papillomatous. They are rarely painful, sometimes cause itching, and are found mainly on the trunk but can appear on any body part. Dermal neurofibromas rarely, if at all, undergo sarcomatous change, but it is prudent to remove rapidly enlarging or painful lesions.
A less common form of peripheral neurofibroma is the nodular neurofibroma, present in about 5% of Nf1 patients. These develop subcutaneously on the major peripheral nerve trunks, and have a much firmer consistency and more defined margins. They are palpated under the skin or found in deeper parts of the body. They often give rise to neurological symptoms such as sensorimotor deficit due to pressure on the peripheral nerves and are a source of pain.
Pathology of neurofibromas.
Lisch nodules
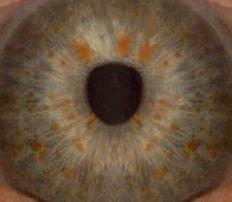
These are harmless, asymptomatic iris hamartomas. They don’t disturb the vision. They appear on slit-lamp examination as being smooth, dome-shaped, and usually light brown in colour. They typically appear between 5 and 10 years of age. Lisch nodules are pathognomonic of NF1 and should be distinguished from iris nevi, which are observed in the general population.
"Lisch Nodules in Neurofibromatosis Type 1, 1991:"http://www.nejm.org/doi/full/10.1056/NEJM199105023241807"
Macrocephaly of unknown aetiology is seen in about 50% of people with Nf1. A child with Nf1 and macrocephaly, therefore, does not need to be investigated unless there are other symptoms or signs suggestive of intracranial pathology, or when serial measurements of head circumference show progressive enlargement.
Approximately 30% of Nf1 patients are at or below the 3rd percentile in height. When compared with unaffected siblings, Nf1 patients are usually 7-8 centimetres shorter. The cause of short stature is unknown.
Multiple juvenile xanthogranulomas may develop in 1%2% of children with Nf1 predominantly on the trunk, head and extremities. They present in early childhood and disappear with age. There is some suggestion in the literature that Nf1 patients with xanthogranuloma are at increased risk for developing juvenile chronic myeloid leukaemia, but this may well just be a coincidental occur rence of rare Nf1 features due to report bias.
Angiomas (Campbell de Morgan spots), mostly on the trunk and thighs, are more common in Nf1 patients than in the general population irrespective of age.
Hypertelorism and thoracic abnormalities including pectus excavatum and pectus carinatum have been documented in 63% and 37.6% of patients in one series, respectively. Documentation of such features may be a helpful aid in assessing the possibility of a diagnosis of Nf1 in children with only CAL spots.
A Noonan syndrome phenotype occurs in approximately 12% of individuals with NF1 (Neurofibromatosis/Noonan phenotype: a variable feature of type 1 neurofibromatosis, 1996). The features may include ocular hypertelorism, down-slanting palpebral fissures, low-set ears, webbed neck, and pulmonic stenosis. Relatives of such individuals who are affected with NF1 may or may not have concomitant features of Noonan syndrome. The NF1-Noonan phenotype appears to have a variety of causes, including the occurrence of two different relatively common autosomal dominant mutations in some families and segregation as an NF1 variant in others.
LEOPARD syndrome , which resembles Noonan syndrome but is distinguished by the presence of multiple lentigines, sensorineural deafness, and a higher prevalence of hypertrophic cardiomyopathy and electrocardiographic alterations, is also caused by PTPN11 mutations in most cases. However, one individual with LEOPARD syndrome and some features of NF1 has been reported to have an NF1 mutation (Neurofibromatosis type I gene mutation in a patient with features of LEOPARD syndrome, 1996).
Pregnancy. Although most pregnancies in women with NF1 are normal, serious complications can occur (Neurofibromatosis type 1 and pregnancy, 1996).
Many women with NF1 experience a rapid increase in the number and size of neurofibromas during pregnancy. Hypertension may first become symptomatic or, if preexisting, may be greatly exacerbated during pregnancy.
Large pelvic or genital neurofibromas can complicate delivery, and cesarean section appears to be necessary more often in pregnant women with NF1 than in other women.
DIAGNOSIS
The diagnosis of
NF1 is usually based on: Family history, physical examination, clinical signs,
MRI and genetic testing.
The clinical diagnosis is generally based on the criteria developed by the
NIH in 1988 (
Guidelines for the diagnosis and management of individuals with neurofibromatosis 1, 2007;
Neurofibromatosis type 1 revisited, 2009), the diagnosis is valid in an individual that shows two or more of the following features:
- Six or more café au lait macules over 5 mm in greatest diameter in prepuberal individuals and over 15 mm in greatest diameter in postpuberal individuals
- Two or more neurofibromas of any type or one plexiform neurofibroma
- Freckling in the axillary or inguinal regions
- Optic glioma
- Two or more Lisch nodules (iris hamartomas)
- A distinctive osseous lesion such as sphenoid dysplasia or tibial pseudarthrosis
- A first-degree relative (parent, sib, or offspring) with NF1 as defined by the above criteria.
The NIH criteria are both highly specific and highly sensitive for adults with NF1.
Diagnosis in children is a bit harder because:
- Only approximately half of children with NF1 and no known family history of NF meet the NIH criteria for diagnosis by age one year, but almost all do by age eight years because many features of NF1 increase in frequency with age. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children, 2000.
- Children who have inherited NF1 from an affected parent can usually be identified within the first year of life because diagnosis requires just one feature in addition to a positive family history. This feature is usually multiple café au lait spots, which develop in infancy in more than 95% of individuals with NF1.
- Young children with multiple café au lait spots and no other NF1 features whose parents do not show signs of NF1 on careful physical and ophthalmologic examination should be strongly suspected of having NF1 and followed clinically as though they do.
- A definite diagnosis of NF1 can be made in most of these children by age four years using the NIH criteria.
Many genetic tests can be used for the diagnosis:
- Sequence analysis of mRNA and genomic DNA which detects : nonsense mutations, missense mutations, splicing mutations and small isìnsertions or deletions in NF1 gene in almost 90% of patients with clinical diagnosis;
- Deletion/duplication analysis (FISH) which detects large deletions (whole gene) in almast 5% of patients with clinical diagnosis;
- Deletion/duplication analysis (MLPA) which detects small intragenic deletions or duplications in almost 1% of patients with clinical analysis;
- Cytogenetic analysis which detects large scale rearrangements in less than 1% of patients with a clinical diagnosis.
Magnetic Resonance Imaging is useful in children to visualize the so called “unidentified bright objects” (UBOs) on brain scan in at least 60% of children with NF1, but their clinical significance is uncertain.
PATHOGENESIS AND GENTICS
NF1 is an autosomally dominantly inherited disorder, which is caused by mutations of the NF1 gene. NF1 encodes the protein neurofibromin, which appears to be a negative regulator of the ras signal transduction pathway. Tumor suppressors normally prevent cells from growing and dividing too rapidly or in an uncontrolled way. This protein appears to prevent cell overgrowth by turning off another protein (called ras) that stimulates cell growth and division.

The NF1 gene is located in chromosome 17q11.2 and it spans over 3.5 kb of genomic DNA (A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations, 1990 ; Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients, 1990).
The NF1 gene is ubiquitously expressed in human, resulting in a 11-13 kb NF1 mRNA and many alternatively spliced variants (A potential role for NF1 mRNA editing in the pathogenesis of NF1 tumors, 1997). The NF1 gene contains 60 exons that are organized into four clusters which are separated by four large introns.

The mutation rate of the NF1 gene is one of the highest known to occur in the human genome, and about 50% of all NF1 patients lack a family history of the disease.
In general, mutational analysis of the NF1 gene is complex due to the large size of the gene, the existence of pseudogenes and the great diversity of the lesions.
A diversity of the mutations affecting the transcript or untranslated regions (UTR) has been described, including e.g. chromosomal abnormalities, deletions, insertions and spot mutations.
In addition to germ line mutations, somatic mutations or LOH have also been shown in many NF1-associated malignancies as well as in cancers of non-NF1 patients.
LOH has also been described in some of the benign neurofibromas. Furthermore, a few cases of somatic mosaicism have been described at the molecular level.
NF1 mRNA is an ubiquitously expressed 11-13 kb transcript, which encodes a protein of 2818 amino acids.
The expression of different NF1 transcripts is specific for different tissues, developmental stage, or modulated by extracellular factors.
Aberrations in NF1 RNA processing might contribute to the marked variation in the clinical picture of NF1. The mechanisms of the NF1 RNA processing known so far are summarized in figure below.

Patients with deletion of the whole gene have been shown to have a recognisable dysmorphic phenotype, which changes with age, and they also have more neurofibromas (including spinal neurofibromas), learning difficulties and certain malignant tumours. The penetrance of the gene has been shown to be almost 100% by the age of five years, but its expressivity is highly variable. About 50% of all patients with NF1 represent new mutations, predominantly ones derived from paternal meiosis. The genetic fitness of NF1, i.e. the ability to propagate, has been shown to be reduced to about half of that expected.
There are some cancers associated with the NF1 gene. In rare cases, inactivation of one copy of the NF1 gene in each cell increases the risk of developing juvenile myelomonocytic leukemia (JMML). Juvenile myelomonocytic leukemia is cancer of blood-forming tissue that usually occurs in children younger than 2.
Neurofibromin is ubiquitously expressed, including nerve cells and specialized cells called oligodendrocytes and Schwann cells that surround nerves, but the protein levels vary in different tissues, and in developmental or functional states.
A portion of the coding sequence of the Nf1 gene (about 13%) shows close homology to the GTPase-activating protein (GAP) family. This region is known as the GAP-related domain (Nf1-GRD). GAP proteins are involved in the regulation of ras, and the presence of this domain supports a tumour suppressor function for Nf1. Some but not all tumours associated with Nf1 show loss of heterozygosity at the Nf1 locus, providing further evidence for a tumour suppressor action. Neurofibromin is involved in control of cell growth and differentiation by at least three possible mechanisms: as an upstream down-regulator of p21 ras, as a downstream effector of p21 ras, and as a link between tubulin and p21 ras.
The NF1 protein has shown to contain six potential phosphorylation sites for serine/threonine kinases and one for tyrosine kinases. The increased phosphorylation level of NF1 protein has been shown to alter the lysosomal degradation rate in melanocytes, and to occur in B lymphocytes in response to IgM-crosslinking, which initiates the redistribution of NF1 protein. The protein kinases phosphorylating the NF1 protein have not been characterized in detail.
The NF1 tumor suppressor is thought to play crucial roles in Ras and cAMP-dependent protein kinase A (PKA)-associated signaling pathways. The NF1 protein has been shown to act as a negative regulator of the p21ras-signaling pathway.
NF1 protein acts as a negative regulator of the Ras. The GRD of the NF1 protein accelerates the switch of active Ras-GTP to an inactive Ras-GDP. The NF1 protein has also been shown to interact with cytoskeleton and to be involved in the adenylyl cyclase / PKA pathway. Growth signals (ligand coupled to receptor) activate guanine nucleotide exchange factors (GEFs), which enables GTP binding to Ras-proteins. Signaling pathways downstream of Ras-GTP include phosphatidylinositol-3-kinase (PI3-kinase), Ral-Rac-Rho-, and raf-MEK-ERK kinase cascades.

Many NF1-deficient tumors have been shown to contain elevated levels of Ras-GTP and/or Ras-dependent signaling pathways.
Recently, several studies have demonstrated that in some cell types the NF1 protein may regulate the cAMP / PKA –pathway rather than Ras. Even early studies have shown that the NF1 protein can complement the function of IRA-proteins, which regulate Ras / cAMP pathway in yeast cells.
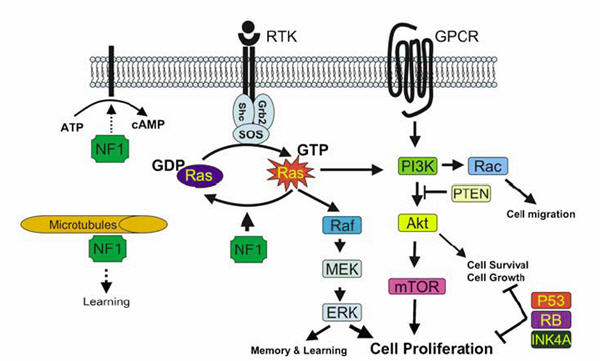
An additional functional specturum for the NF1 protein might be achieved with alternative splicing, phosphorylation, or regulatory molecules. Thus, it is not surprising that the NF1 protein seems to have pleiotrophic effects on cell behavior.
The GTPase activities of NF1 protein and GAP are differentially regulated: dodecyl maltoside and lipids (e.g. arachidonic acid, phosphatidic acid, stearic acid, oleic acid, phosphatidylinositol-4,5-bisphosphate, some n-6 and n-3 polyunsaturated fatty acids) inhibit the activity of NF1 protein more than GAP. The association of NF1 protein with tubulin has also been shown to inhibit the GTPase stimulating activity of NF1 protein. Furthermore, the functional properties of NF1 protein isoforms differ markedly. The NF1 protein isoforms I-IV and 9br contain both GRD and microtubule-binding regions. The insertion of 21 amino acids in the GRD of type II and IV NF1 isoforms weakens the GTPase stimulating activity. The N-isoforms of the NF1 protein lack both GRD and tubulin-binding domains. In contrast, the editing of the NF1 mRNA disrupts GRD, but retains the tubulin binding regions of the the protein.
Subcellular localization of the NF1 tumor suppressor and its co-localization with other molecules have given clues of its putative functions. The NF1 protein has been shown to associate with the microtubular and microfilamentous cytoskeleton, and even sequentially with both within the same cell. Furthermore, the interaction between NF1 protein and syndecan and paxillin (the scaffold for molecules associating with focal adhesions) has been suggested. In addition, NF1 protein has been found in the mitochondria and nucleus, and in association with both the membrane and cytosolic fractions.
COMPLICATIONS
Neurological complications
Cognitive impairment.
The deficit in neurocognitive complications are most often reported in NF1. The intellectual impairment in Nf1 presents in childhood as learning difficulties and is relatively mild and non-progressive.
The learning deficits in children with NF1 may include face-spatial deficits and face motor and speech disorders. In addition to specific deficits, nonverbal and verbal, seen in 30-60% of children with NF1 are common deficits of motor coordination, fine and coarse.
Hyperintense brain lesions on T2-weighted MR images.
High-signal-intensity lesions on T2-weighted MRI of the brain are a frequent finding in individuals with Nf1. These are well-circumscribed, round to ovular, nonenhancing hyperintense lesions that usually do not produce mass effect. They are referred to as “unidentified bright objects” (UBOs) and are most commonly found in the basal ganglia, especially in the globus pallidus, internal capsule, thalamus, cerebellum, and brain stem regions. They are generally asymmetrical and occur in about 60% of children young adults with Nf1 who undergo MRI, but they tend to disappear in adulthood and are seldom observed in patients over 30 years of age. They do not cause overt neurological symptoms.
Their pathological correlation is yet debated.
Unidentified bright objects on brain MRI in children as a diagnostic criterion for neurofibromatosis type 1, 2008.
UBOs show signs of abnormal myelin structure on diffusion-weighted MRI and correspond pathologically to areas of spongiform myelinopathy. They may disappear with age and are less common in adults than in children with NF1.
Some studies have suggested that the presence, number, volume, or location of UBOs correlates with learning disabilities in children with NF1, but the findings have not been consistent across investigations.
Brain tumours
Gliomas are a well-recognised complication of Nf1 and may arise in all parts of the brain, although the sites of predilection are the optic pathway, brain stem and cerebellum. Medulloblastomas and ependymomas have also been described in Nf1.
Optic pathways gliomas (OPG) are pilocytic astrocytomas which cause expansion of the optic nerve. OPG may spread posteriorly to the optic chiasm and then either into the contralateral optic nerve, thalami or internal capsule; they may even cause obstruction of the cerebrospinal fluid (CSF) circulation leading to hydrocephalus. The peak-age for developing an OPG in Nf1 is 4.9 years and symptomatic tumours seldom arise after the age of 6 years.
The optic nerve gliomas are found in approximately 15% of children with NF1 and usually appear in the first decade of life. They have a benign course and only a third to half of patients with gliomas of the optic nerve, associated with NF1, the symptoms manifest. In rare instances spontaneous involution has been documented.
Symptomatic patients complain of deterioration in visual acuity and loss of vision in the peripheral fields. Ophthalmologic examination may reveal impaired colour vision, retinal vein tortuosity, optic atrophy,strabismus or pupillary abnormalities.
Chemotherapy (carboplatin and vincristine) is the first choice treatment because of less side effects; radiotherapy is initially more effective but does not influence the long-term outcome. Surgery takes a role only with those cases having symptomatic unilateral lesions.
The frequency of brain stem and cerebellar tumours is greater in Nf1 than in the general population. These are mostly low-grade gliomas, but are sometimes medulloblastomas or, in the brain stem, ependymomas. Approximately 50% of patients may show clinical or radiological progression at some stage and some may experience symptoms or neurological deficits. These tumours seem, however, to have a more indolent course in Nf1 as compared to when they occur in isolation. In a few patients the clinical course is extremely aggressive.
Intraspinal, intramedullary and paravertebral tumours.
Intrinsic low-grade or atypical gliomas and ependymomas of the spinal cord have been occasionally reported in Nf1 patients, although they are more common in Nf2. They usually present with impairment of pain and temperature sensation, reflex loss at the level of the tumour, sphincter disturbances or progressive paraparesis. They may be only slowly progressive but generally have a poor outcome.
Seizures and epilepsy.
The occurrence of a seizure in an Nf1 patient may well signal the existence of a recognised complication of Nf1 such as tumours, hydrocephalus or cerebrovascular disease, but may, as for seizures in the general population, be idiopathic or consequential to non-Nf-related pathology. There is increased awareness that seizures may be relatively uncommon in Nf1 and not fully explained by underlying CNS lesions.
Headache
There is some suggestion in the literature and it is the experience of those who examine large numbers of Nf1 patients that some individuals with Nf1, especially children, may experience migraine headaches, characterised by steady or throbbing cephalalgia, nausea or abdominal pain.
Aqueductal stenosis.
Hydrocephalus due to aqueduct stenosis is a true, albeit rare, complication of Nf1. In some cases the pathologic basis consists of non-progressive proliferation of subependymal glial cells around the
acqueduct. When symptomatic it requires immediate neurosurgical intervention by shunting.
Cerebrovascular disease
There is an increasing number of reports in the literature on intrinsic abnormalities of the intracranial vasculature in association with Nf1. However, the assumption of increased prevalence of this pathology in Nf1 has to be carefully interpreted since cerebrovascular disease is common in the community at large.
Usually, presentation is with subarachnoid haemorrhage or as an incidental finding, but a miscellany of symptoms is not uncommon.
Neurofibromatous neuropathy
This neuropathy is a rare manifestation of neurofibromatosis: it presents itself as a form of distal, symmetric, clinically characterized by early development of a large number of subcutaneous neurofibromas. The neurological deficit was due to accumulation of multiple peripheral neurofibromata on the nerve roots.
In contrast with neuropathy associated with neurofibromatosis type 2 (including 10% of neurofibromatosis and is characterized by bilateral acoustic neuromas acoustics and a parent with a similar form), neurofibromatosis type 1 neuropathy is accompanied by a sensory deficit without neurophysiological or clinical deterioration.
Neurofibromatous neuropathy in neurofibromatosis 1 (NF1), 2004.
Multiple sclerosis
An increased frequency of multiple sclerosis has been reported in Nf1 patients. Mutations in the oligodendrocyte-myelin glycoprotein (OMGP) gene which predispose to MS and is embedded within the Nf1 gene were postulated but not yet detected.
Malignant peripheral nerve tumours.
Patients with neurofibromatosis have a 10% risk of developing aggressive spindle cell sarcoma, malignant peripheral nerve sheath tumours (MPNST). These tumors arise from plexiform neurofibromas, although in 36% of these malignancies is not described an earlier plexiform neurofibroma.
These may occur at any site in the peripheral nervous system but are most common within plexiform neurofibromas. In approximately 10% of patients they develop at a site that has been previously irradiated for another neoplasm. Patients with MPNST in general have pain and neurological deficits.
The prognosis is usually poor and the tumours tend to occur in younger subjects
Plexiform neurofibromas
The second most common Nf1 complication, after learning difficulties, is plexiform neurofibroma (PNF). The plexiform neurofibromas differ from focal cutaneous neurofibromas, that originate from many nerve bundles, and tend to grow along the length of the nerve. These lesions typically are present at birth and continue to appear until late adolescence and early part of adulthood. They occur in approximately 30% of patients with neurofibromatosis.

They usually present as large subcutaneous swellings, soft in consistency, with ill-defined margins, varying from a few centimetres in diameter to a whole area of the body. Much less frequently, PNFs are nodular with nerve trunks developing discrete nodular tumours. The overlying skin is often hyperpigmented and hypertrophied or shows excessive hair growth. This often produces disfiguring, localised gigantism causing psychological problems.
They are divided into 3 categories: surface, displacing, invasive.
Orthopaedic complications
Bone abnormalities are common in Nf1 and usually arise from intrinsic abnormalities of the skeletal system, from bony overgrowth or destruction caused by underlying PNF.
Scoliosis is present in 10% of Nf1 patients and is of two main varieties of curve:
- Dystrophic scoliosis involves 4-6 segments and causes severe wedging and rotation of the vertebral bodies, ribthinning and scalloping. This form of scoliosis usually appears early in childhood, is rapidly progressive, is uncontrolled by bracing, and requires early fusion;
- Idiopathic scoliosis may be convex to the left or to the right, and can be managed in the same way as conventional idiopathic scoliosis by an instrumented posterior fusion.
Pseudarthrosis of the long bones (usually of the tibia or fibula), due to a congenital defect of bone formation, is seen in 2% of Nf1 patients. Bowing, constriction or a defect in the affected long bone may present at birth. Spontaneous recurrent fractures or fractures following minor trauma manifest in early childhood and usually do not heal spontaneously. Other congenital deformities may be also present. Prognosis varies largely and several surgical techniques have been employed. In severe cases amputation of the affected limb may be needed.
Ophthalmological complications
Besides
OPGs, ophthalmological complications include:
- Neurofibromas involving the eyelids or the orbit,
- Sphenoid wing dysplasia often associated with disruption of the posterior, superior wall of the orbit leading in turn to herniation of the temporal lobe into the orbit and pulsating exophthalmos,
- Idiopathic congenital ptosis (sometimes referred to as part of the so-called Noonanoid phenotype),
- Congenital or acquired glaucoma (usually unilateral),
- Choroidal hamartomas,
- Diffuse or nodular enlargement of the corneal nerves (not pathognomonic of the disease).
General medical complications
The manifestations of NF1 include cardio-vascular diseases, congenital heart, vascular disease and hypertension.
Blood pressure and cardiovascular involvement in children with neurofibromatosis type1, 2004.
The coronary heart disease occur more often in NF1 than you find in the general population. The pulmonary artery stenosis accounts for 25% of cardiac malformations. All children must be subjected to listening to the heart and blood pressure.
Vascular disease in NF1 are represented by stenosis, aneurysms and arteriovenous malformations, which represent the second leading cause of death in these patients. The rule of vascular disease affecting the arterial system and the renal artery stenosis is the most common manifestation, present in 1% of patients with NF1. Renal arteriography is indicated in every patient with hypertension and NF1. Cerebrovascular diseases, especially in younger patients, are the result of stenosis and occlusions and are diagnosed in children with a clinical question of fatigue, involuntary movements, headache or seizures secondary to ischemia. Physiologically, vascular lesions show dysplasia fibromuscular intimal thickening and proliferation of Schwann cells, without atherosclerosis.
Hypertension: is significantly associated with NF1: blood pressure should be checked annually with a limit <140/90 mmHg. Renal artery stenosis is the most common situation. However they can be found coarctation of the aorta, and pheochromocytoma.
Pheochromocytoma is found with a frequency from 0.1 to 5.7%. Most (90%) are benign and typically occur in the adult population. However, the risk of malignancy in any patient with hypertension, especially if paroxysmal type, or with symptoms of excess catecholamines, such as headache, sweating, palpitations or anxiety, should be measured within 24 hours, the urinary concentration total and fractionated catecholamines and their metabolites. Only after the presence of a pheochromocytoma has been confirmed biologically, should be performed MRI to locate the tumor. There is an association between pheochromocytoma and carcinoid tumors, usually in the duodenum, so the presence of one should prompt the clinician to look for another.
Respiratory problems are infrequent in Nf1 but intrapulmonary neurofibromas or significant scoliosis may cause lung disease. Carcinoid tumours, gastrointestinal neurofibromas, gangliocytic paragangliomas and ganglioneuromas have been, albeit rarely, reported in Nf1. They may cause pain, dyspepsia, haematemesis and melaena, abdominal distension, discomfort or constipation, and they must be taken into high consideration when dealing with gastrointestinal problems in Nf1 patients.
Malignancy
Malignant tumours occur four-times more frequently in Nf1 patients than in the general population. Cross-sectional and retrospective cohort studies give credible evidence for the validity of the association of Nf1 with some central nervous system tumours (especially glioma of the optic pathway), neurofibrosarcoma and phaeochromocytoma whose relative risk is surely high, but the absolute risk is low. The common adult cancers, however, do not seem to be associated with Nf1.
By contrast, certain leukaemias (especially non-lymphocytic leukaemias of childhood) are validly associated tumours. The prognosis in Nf1 malignancies is not different from that of non-Nf1 individuals with the same tumour and stage and, therefore, there are no reasons to modify therapy just because the patient has Nf1.
THERAPY
After the diagnosis all the clinical aspects should be evaluated and treated (if necessary) to prevent the complications that may occur.
To prevent complications, reduce comorbidities and improve quality of life annual consultation and physical examination are required. The patient should be examined by:
- A neurologist to provide information about changes in neurological status
- A neurosurgeon to identify and treat spinal cord or brain tumors.
- An opthalmologist to get information’s about visual acuity, fields defects or appearance of lisch nodules.
- An orthopedist to evaluate the bone related abnormalities.
Blood pressure should be checked frequently and hypertension should be treated promptly if detected.
Every change that may occur in sensory or motor examination (such as incontinence) should be documented and evaluated carefully.
Neurofibromatosis, Type 1 Treatment & Management
To treat malignant peripheral nerve sheat tumors (MPNSTs) that are unresecable chemotherapy can be used (For example farnesyl transferase used in combination with lovastatin reduce growth of MPNSTs).
Surgery can be useful to resect neurofibromas that press on vital structures, obstruct vision, or to resect plexiform neurofibromas which surgical approach could be difficult and in most cases is unsatisfactory because of recurrence due to cell collection in soft tissues.
Treatment of optic gliomas is problematic as they are usually asymptomatic and clinically stable or only very slowly progressive.
Orthopedic intervention can be indicated for rapid progressive scoliosis and for severe bone defects.
Hypertensive patients with renal artery stenosis could require surgical resection and repair or angioplasty.
Plastic surgery consultation is advisable for areas of great cosmetic concern (for example the face).
MRI for follow-up of clinically suspected intracranial tumors and other internal tumors.
No activity restrictions are necessary, except for an individual with specific orthopedic concerns.
NEW THERAPIES
New therapies for cancer, associated with
NF1 can be grouped into:
- Those who seek to deregulate signaling pathways within tumor cells and
- Those who seek to change the stromal component in tumor microenvironment.
In addition, strategies have been proposed based on the correction signal of Ras for the treatment of cognitive deficits in children with NF1.
Against tumor cells
Because neurofibromin functions as an inhibitor of Ras, early studies have focused on inhibitors of Ras. The tipifarnib is an inhibitor of farnesyl protein transferase, which inhibits farnesyl and geranilgeranilazione of Ras, required for its transfer to the cell membrane and subsequent activation. Have recently completed Phase 1 trials with tipifarnib, performed in children with refractory solid tumors or with plexiform neurofibromas in NF1 course, the drug was well tolerated in children and adults.
Because neurofibromin regulates mTOR signaling, the use of rapamycin and its analogs should be considered in the treatment of cancer in individuals with NF1. Rapamycin was initially described as an immunosuppressive drug that binds to its target FKBP12, an inhibitor of mTOR signaling. Interest in the use of rapamycin has been raised recently by the description of rapamycin by the mouth causes a regression of giant-cell astrocytoma subependymal, in a small number of patients with tuberous sclerosis.
Towards the stromal component
Antihistamine agents such as ketotifen, although not with any favorable effect on the treatment of plexiform neurofibromas, are used because they would lead to an attenuation of subjective symptoms.
The plexiform neurofibromas maintain an abundant blood supply, suggesting that therapeutic agents that act on tumor vasculature, can be effective.
It was first used to interferon α, but the practical implementation was disappointing. The AZD2171, which inhibits the receptor tyrosine kinase is known as angiogenesis inhibitor, this substance and another inhibitor of angiogenesis (thalidomide) are probably effective in the treatment of peripheral nerve sheath tumors. The AZD2171 is used in phase 1 and plexiform neurofibromas in patients with spinal neurofibromas.
The pirfenidone, antifibrotic a substance that helps to reduce the activity of cytokines released from fibroblasts in the neighborhood of neurofibromas, renders it incapable of acting to the cellular network support (fibroblasts, mast cells and others). Phase II trial of pirfenidone in adults with neurofibromatosis type 1, 2006.
G. Bartolozzi. NEUROFIBROMATOSI TIPO I, 2009. Medico e Bambino pagine elettroniche 2009
STATIN THERAPY IN NEUROFIBROMATOSIS TYPE I
Given the role of neurofibromin in stimulating the conversion of Ras-GTP to Ras-GDP, proteins involved in the Ras signal transduction pathway have been identified as potential therapeutic targets.
The Nf1 +/- mouse model has been used to model learning disabilities. The phenotype is rescued by genetic crosses that reduce Ras-GTP in the brain, as well as by treatment with a farnesyl transferase inhibitor, which blocks Ras binding to the cell membrane. The learning deficits associated with NF1 may be caused by excessive Ras activity, which leads to impairments in long-term potentiation caused by increased GABA-mediated inhibition.
Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1, 2002.
GABA release impairment
Learning triggers interneuronal Ras signaling leading to increased GABA release. MAPKdependent phosphorylation of synapsin I (SynI) plays a critical role in GABA release. Wild-type NF1 restricts the increase in GABA release within an appropriate range that modulates learning. In Nf1 mutants, reduced NF1 activity leads to abnormal hyperactivation of Ras signaling in inhibitory interneurons during learning, resulting in abnormally high GABA release. This increased activity-dependent GABA release shifts the balance between excitatory and inhibitory processes in neuronetworks of the mutant mice and impairs synaptic plasticity needed for learning and memory.
Molecular and cellular mechanisms of learning disabilities: a focus on NF1, 2010.
So evidence from mouse studies suggests that an overabundance of active Ras results in abnormal nerve-cell responses in the brain.

Statins are inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase, broadly used for the reduction of serum cholesterol. As statins inhibit the initial enzyme of the mevalonate pathway, they also reduce prenylation and farnesylation of signalling molecules, such as Ras and Ras-related proteins.
Statins lower cholesterol by blocking the effects of certain fats. Because Ras requires fat to function, a decrease of fat corresponds to a reduced activity of the protein. Experiments on mice with the NF1 mutation (and with the same symptoms as humans: attention deficit, learning problems and poor physical coordination) have confirmed that the drug enables the animals to achieve performance similar to those of normal control subjects .
It was demonstrated that lovastatin rescues the cognitive defect, presumably by virtue of its effect on membrane binding of Ras. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1, 2005
One negative statin trial has recently been reported. Effect of simvastatin on cognitive functioning in children with neurofibromatosis type 1: a randomized controlled trial, 2008
Skeletal problems and osteoporosis occur in up to 50% affected neurofibromatosis type 1 (NF1) humans. Inactivation of neurofibromin results in deregulation of Ras signal transduction. Little is known of bone biology in humans with NF1. Loss-of-function of Nf1 gene was associated with altered bone homeostasis and Ras signal transduction. Neurofibromin and its inactivation of Ras are prerequisites for osteoblast functioning, 2005.
It was demostrated that skeletal dysplasia may thus be another target of lovastatin therapy in clinical trials for patients with NF1.
Modelling neurofibromatosis type 1 tibial dysplasia and its treatment with lovastatin, 2008
Multiple roles for neurofibromin in skeletal development and growth, 2007
Improvement of bone healing in children with tibial dysplasia would be welcomed, given the enormous difficulty in the management of this complication and the severe associated morbidity. Whether statin treatment will have other benefits related to bone mineral density remains to be determined. Moreover, there is a possibility that these findings will have an impact on the management of bone disorders in non-neurofibromatosis-affected individuals as well. When patient-advocates argue to increase funding for research on neurofibromatosis, one of the potential benefits cited is that this work will shed light on common disorders such as cancer, learning disabilities, and osteoporosis. The NF1 gene clearly plays a role in normal bone development and metabolism, and there is indeed a possibility that pathogenetic and therapeutic insights gained from this rare disorder may someday benefit a much larger population.
Statins, bone, and neurofibromatosis type 1, 2008