DEFINITION
Mitofusin-2 is a protein that in humans is encoded by the MFN2 gene. Mitofusins are GTPases embedded in the outer membrane of the mitochondria. In mammals MFN1 and MFN2 are essential for mitochondrial fusion.
THE GENE
The gene MFN2, also known as HSG, MARF, CMT2A, CPRP1, CMT2A2, encodes a mitochondrial membrane protein (Mitofusin-2) that participates in mitochondrial fusion and contributes to the maintenance and operation of the mitochondrial network. Mutations in this gene cause Charcot-Marie-Tooth disease type 2A2, and hereditary motor and sensory neuropathy VI, which are both disorders of the peripheral nervous system. This protein is involved in the regulation of vascular smooth muscle cell proliferation, and it may play a role in the pathophysiology of obesity. Defects in this gene have also been associated with early-onset stroke.


By radiation hybrid analysis and by examining a human-rodent hybrid panel, the MFN2 gene was mapped to chromosome 1(Prediction of the coding sequences of unidentified human genes. VI. The coding sequences of 80 new genes (KIAA0201-KIAA0280) deduced by analysis of cDNA clones from cell line KG-1 and brain, 1996). The MFN2 gene maps to chromosome 1p36.2 (Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A, 2004), about 1.65 Mb centromeric to the KIF1B gene (605995), which is mutant in Charcot-Marie-Tooth disease-2A1 (CMT2A1; 118210). Moreover, the MFN2 gene contains 19 exons (Phenotypic spectrum of MFN2 mutations in the Spanish population, 2010).

CHEMICAL STRUCTURE AND IMAGES
Mitofusin-2 is one of the two mitofusin proteins also required for mitochondrial fusion. Mfn1 and Mfn2 are conserved integral outer mitochondrial membrane proteins, each consisting of a large GTPase domain and two heptad repeats, or putative coil-coiled domains, all of which face the cytoplasm (Structural Basis of Mitochondrial Tethering by Mitofusin Complexes, 2004).

Protein Aminoacids Percentage


CELLULAR FUNCTIONS
Mitofusin 2 is a nuclear encoded, dynamin-like transmembrane GTPase, whose normal function is helping determine the morphology of mitochondria and tethering the endoplasmic reticulum to mitochondria. Mitofusin 2 is made in many types of cells and tissues, including muscles, the spinal cord, and nerves that connect the brain and spinal cord to muscles (peripheral nerves). Within cells, mitofusin 2 is found in the outer membrane that surrounds mitochondria.
Mitochondria are dynamic structures that undergo changes in morphology through processes called fission (splitting into smaller pieces) and fusion (combining pieces). These changes in morphology are necessary for mitochondria to function properly. Mitofusin 2 helps to regulate the morphology of mitochondria by controlling the fusion process. Mitochondrial fusion and fission are mediated by several GTPases in the outer mitochondrial membrane, notably mitofusin-2 (Mfn-2), which promotes fusion, and dynamin-related protein (Drp-1), which promotes fission.

MFN1 and MFN2 mediate outer membrane fusion while OPA1 is involved in inner membrane fusion.
Mitochondrial fusion is essential for embryonic development. Knockout mice for either MFN1 or MFN2 have fusion deficits and die midgestation. MFN2 knockout mice die at embryonic day 11.5 due to a defect in the giant cell layer of the placenta (Mitochondria: dynamic organelles in disease, aging, and development,2006).
Fusion is also important for mitochondrial transport and localization in neuronal processes. Conditional MFN2 knockout mice show degeneration in the Purkinje cells of the cerebellum, as well as improperly localized mitochondria in the dendrites. MFN2 also associates with the MIRO-Milton complex which links the mitochondria to the kinesin motor (Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration, 2012).
Regulation
MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2.
Mitochondria are cellular organelles constantly subject to highly dynamic processes, such as fission and fusion. The protein MITOL, localized to the outer mitochondrial membrane, regulates mitochondrial dynamics, interacting with the protein Mfn2. Mfn2 is enriched at the junction between the endoplasmic reticulum (ER) and mitochondria, which is known as the mitochondria-associated ER membrane (MAM), which is essential for many cellular functions. In this study, the authors have demonstrated the role of the protein MITOL about the interaction of the endoplasmic reticulum and mitochondria, by modulating the Mfn2 activity via K192 ubiquitination. Indeed, MITOL knockdown modifies Mfn2 localization, MAM function and their their regulation by MITOL. MITOL could be involved in the pathogenesis of neurodegenerative diseases, such as CMT2A, through Mfn2 regulation. Future studies about Mfn2 role, MAM domain and especially about their regulation mediated by MITOL could contribute greatly to our understanding of the mechanism of CMT2a (MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2, 2013).
CHARCOT-MARIE-TOOTH (CMT) DISEASE SUBTYPE CMT2A
Charcot-Marie-Tooth disease is a group of disorders passed down through families that affect the peripheral nerves.
It is a hereditary motor and sensory neuropathy (HMSN) and peroneal muscular atrophy (PMA) characterised by progressive loss of muscle tissue and touch sensation across various parts of the body. Currently incurable, this disease is one of the most common inherited neurological disorders.
CMT is a result of genetic mutations in a number of genes. Based on the affected gene, CMT can be categorized into types and subtypes. Mutations in the mitofusin 2 gene (MFN2) result in the axonal subtype CMT2A.
Disease characteristics
Charcot-Marie-Tooth hereditary neuropathy type 2A (CMT2A) is a classic axonal peripheral sensorimotor neuropathy characterized by earlier and more severe involvement of the lower extremities than the upper extremities, distal upper-extremity involvement as the neuropathy progresses, more prominent motor deficits than sensory deficits, and normal (>42 m/s) or only slightly decreased nerve conduction velocities (NCVs). Postural tremor is common. Most affected individuals develop symptoms in the first or second decade. It has recently been suggested that CMT2A represents more than 90% of the severe dominant CMT2 cases. However, milder late-onset cases and unusual presentations have also been described (Charcot-Marie-Tooth Neuropathy Type 2A, 2005).
Epidemiology
The overall prevalence of hereditary neuropathies is estimated at approximately 3:10,000 population. About 30% of these individuals (*1:10,000*) may have CMT2. CMT 2A2 affects around 19-23% of patients suffering from CMT2 type axonal neuropathy (inherited axonal neuropathy transmitted as autosomal dominant characteristic), thus representing the most frequent subtype of CMT2 type diseases. A study in 2006 published by the scientific journal Brain reported the percentage of affected patients as up to 33% (Genetic epidemiology of Charcot-Marie-Tooth disease, 2012).
Geographical prevalence
CMT2A represented 3.4%-16% of all CMT families in Norway and Spain respectively (An investigation of 232 Norwegian CMT families, 2010 and Phenotypic spectrum of MFN2 mutations in the Spanish population, 2010). In a large study of German individuals with a CMT2 phenotype (776), the following percentages were found: 11% had CMTX1, 8% had CMT2A and 6% had the rare giant axonal neuropathy. Among those with CMT2, 35% had a genetic diagnosis (Charcot-Marie-Tooth disease: frequency of genetic subtypes in a German neuromuscular center population, 2013).
Inheritance
The Mitofusin 2 gene mutation responsible for axonal neuropathy is mainly transmitted as an autosomal dominant characteristic, i.e. the mutation carrier is a parent who will have a 50% chance of passing on the disease to the child. In some rare cases the genetic transmission is homozygous recessive, i.e. both parents present with a MF2 gene mutation. In such cases the child will have a 25% chance of being healthy, a 25% chance of receiving both mutations and a 50% chance of receiving one mutation. Parents and children carrying one mutation can be either asymptomatic or presenting with mild neuropathy. There are also compound trans-heterozygous mutations associated with a pattern of autosomal recessive transmission. These often determine a more severe, early-onset phenotype of the disease.
CMT 2A2 can also result from de novo mutations, with no precedent family history.
Penetrance of the disease is variable, thus meaning that within the same families multiple occurrences can present with different clinical statuses in terms of characteristics and severity (Autosomal-recessive Charcot-Marie-Tooth diseases, 2005).
Mutation
The presence of some of those characteristics can be related to specific Mitofusin 2 mutations. To date, scientists have discovered more than 80 mutations responsible for the disease (Novel mutation of the mitofusin 2 gene in a family with Charcot-Marie-Tooth disease type 2, 2014).
Researchers have identified approximately 50 MFN2 gene mutations that cause a form of Charcot-Marie-Tooth disease known as type 2A. Almost all of these mutations change single protein building blocks (amino acids) in mitofusin 2. These genetic changes alter a critical region in mitofusin 2, and the protein cannot function properly. A few mutations create a premature stop signal in the instructions for making mitofusin 2. As a result, no protein is produced, or an abnormally small protein is made.
Several MFN2 gene mutations cause a variant of type 2A Charcot-Marie-Tooth disease that affects vision. (This variant is also called hereditary motor and sensory neuropathy VI.) Vision loss is caused by the degeneration of the nerves that carry information from the eyes to the brain (optic atrophy). People with this variant usually experience severe symptoms of Charcot-Marie-Tooth disease that begin before age 10.
It is unclear how MFN2 gene mutations lead to the nerve problems characteristic of type 2A Charcot-Marie-Tooth disease. Researchers suggest that mitochondria cannot fuse properly or move normally within the cell without functional mitofusin 2, which may disrupt the cell's energy supply. Nerve cells may be particularly sensitive to an interrupted supply of energy (Clinical, electrophysiological and pathological findings of a patient with CMT2 due to the p.Ala738Val mitofusin 2 mutation, 2011).
Pathogenesis
Axonal transport
Altered mitochondrial transport has been suggested to be a pathological mechanism of axonal CMT neuropathy
Alterations in mitochondrial dynamics (fission, fusion, and movement) are implicated in many neurodegenerative diseases, from rare genetic disorders such as Charcot-Marie-Tooth disease, to common conditions including Alzheimer's disease. However, the relationship between altered mitochondrial dynamics and neurodegeneration is incompletely understood. Here we show that disease associated MFN2 proteins suppressed both mitochondrial fusion and transport, and produced classic features of segmental axonal degeneration without cell body death, including neurofilament filled swellings, loss of calcium homeostasis, and accumulation of reactive oxygen species. By contrast, depletion of Opa1 suppressed mitochondrial fusion while sparing transport, and did not induce axonal degeneration. Axon degeneration induced by mutant MFN2 proteins correlated with the disruption of the proper mitochondrial positioning within axons, rather than loss of overall mitochondrial movement, or global mitochondrial dysfunction. We also found that augmenting expression of MFN1 rescued the axonal degeneration caused by MFN2 mutants, suggesting a possible therapeutic strategy for Charcot-Marie-Tooth disease. These experiments provide evidence that the ability of mitochondria to sense energy requirements and localize properly within axons is key to maintaining axonal integrity, and may be a common pathway by which disruptions in axonal transport contribute to neurodegeneration (Mitofusin2 Mutations Disrupt Axonal Mitochondrial Positioning and Promote Axon Degeneration, 2012).
Autosomal dominant optic atrophy plus phenotype
We report a large family with optic atrophy beginning in early childhood, associated with axonal neuropathy and mitochondrial myopathy in adult life. The clinical presentation looks like the autosomal dominant optic atrophy 'plus' phenotype linked to OPA1 mutations but is associated with a novel MFN2 missense mutation (c.629A>T, p.D210V). Multiple mitochondrial DNA deletions were found in skeletal muscle and this observation makes MFN2 a novel gene associated with 'mitochondrial DNA breakage' syndrome. Contrary to previous studies in patients with Charcot-Marie-Tooth disease type 2A, fibroblasts carrying the MFN2 mutation present with a respiratory chain deficiency, a fragmentation of the mitochondrial network and a significant reduction of MFN2 protein expression. Furthermore, we show for the first time that impaired mitochondrial fusion is responsible for a deficiency to repair stress-induced mitochondrial DNA damage. It is likely that defect in mitochondrial DNA repair is due to variability in repair protein content across the mitochondrial population and is at least partially responsible for mitochondrial DNA instability (The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy ‘plus’ phenotype, 2012).
Onset
CMT 2A2 onset is variable, ranging from the first decade (2-3 years) up to the fifth decade. Generally, these extreme early onset cases are the most severe.
The clinical history can vary, ranging from early onset cases with severe weakness to asymptomatic cases (up to 25% of all MTF2 gene mutations carriers present with subclinical signs only).
Overall, based on the clinical status we can define:
- An early onset form (within the first decade), which is characterised by a severe phenotype and rapid course including loss of deambulation autonomy (generally within the second decade) leading to an early need for deambulation supports. With regard to neurophysiology, this subtype is associated with severe reduction in the amplitude of the compound muscle action potential (CMAP). The reduction is so significant that in some cases CMAP cannot be detected. There is evidence for this form being associated to the following mutations: L92P, R94W, R94Q,T105M, R364W.}
- A late onset form (after the first decade), which is characterised by a less severe phenotype and a less aggressive course. Neurophysiologic studies show normal or mildly impaired CMAP. There is evidence for this form being associated with the following MFN2 gene mutations: M367T, H165R.
(Early onset severe and late-onset mild Charcot-Marie-Tooth disease with mitofusin 2 (MFN2) mutations, 2006).
Symptoms
In general, the primary symptom is a slow-progression motor and sensory inherited neuropathy. This begins in lower limbs and then expands to upper limbs, causing difficulties in deambulation, weakness in movement and difficulties in hand use.
The range of signs and symptoms includes:
- Polyneuropathy
- distal weakness in hands and feet (affecting mostly legs rather than arms). This results in difficulties in deambulation and can cause complete deambulation impairment;
- hypotonia and reduction or absence of osteotendinous reflexes; frequent fatigue;
- abnormalities in sensitivity.
- Optic atrophy (in most severe cases)
- Pain, cramping
- Tremors
- Hearing loss
- Macrocephaly
- Psychomotor impairment
- Pes cavus, scoliosis
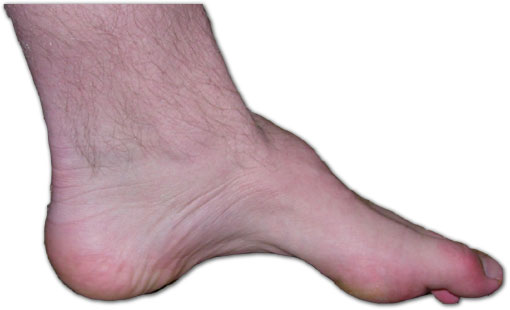
(Charcot-Marie-Tooth disease in Cyprus: epidemiological, clinical and genetic characteristics, 2010).
(Autosomal recessive Charcot-Marie-Tooth disease: from genes to phenotypes, 2013).
(Clinical and genetic study of a large Charcot-Marie-Tooth type 2A family from southern Italy, 2001).
Diagnosis
The diagnosis is established by clinical and molecular genetic findings. MFN2 is the only gene in which mutations are known to cause CMT2A.
Clinical Diagnosis
No specific findings distinguish CMT2A from other types of CMT2. However, in young and severely affected individuals with CMT2, MFN2 mutations are the most frequent cause of disease. Typical findings include those already treated in the ‘Disease Characteristics’ section.
Encephalic NMR can be useful in case of CNS-related symptoms; ophthalmological examination with examination of the fundus oculi and of the field of vision can be useful in case of visual acuity reduction; also, in cases of unclear diagnosis a sural nerve biopsy can be performed to gather additional morphostructural and immunohistochemical information on the peripheral nervous system (Diagnosis of the peripheral hereditary neuropathies and its molecular genetics, 2008).
Neurophysiologic examination
ENG/EMG is performed to study motor and sensory nerve conduction velocities. Those are mildly reduced in axonal neuropathies such as CMT2, with significant reductions in CMAP and SAP. Those results suggest axonal neuropathy (Diagnosis of the peripheral hereditary neuropathies and its molecular genetics, 2008).


Genetic examination
When clinical and neurophysiologic data show evidence compatible with a CMT2A2 form, genetic examination is performed with Mitofusin gene sequencing.
Current genetic test algorithms for Charcot Marie Tooth (CMT) disease are based on family details and comprehensive clinical and neurophysiological data gathered under ideal conditions for clinical assessment (Diagnostic laboratory testing for Charcot Marie Tooth disease (CMT): the spectrum of gene defects in Norwegian patients with CMT and its implications for future genetic test strategies, 2013).
Summary of Molecular Genetic Testing Used in Charcot-Marie-Tooth Neuropathy Type 2A : for this gene (MFN2) the proportion of CMT2A attributed to its mutations is nearly 100%.
| Test Method | Mutations Detected |
| Sequence analysis/mutation scanning | Sequence variants |
| Sequence analysis of select exons | Sequence variants in select exons |
| Deletion/duplication analysis | Exon and whole gene deletions/duplications |
Treatment
To date there is no cure to CMT 2A2, and the only treatment that proved to be capable to improving functional performances (e.g. in prehension or dehambulation) in patients affected by CMT is rehabilitation therapy.
Treatment by a team includes a neurologist, physiatrist, orthopedic surgeon, and physical and occupational therapists.
Essential tools for rehabilitation treatment in CMT patients are orthopaedic supports such as shoes (either normal shoes specifically modified by an orthopaedist or custom-made shoes), insoles and supports to stabilise the ankle and avoid the foot dangling whilst walking. Forearm crutches, canes, wheelchairs may be needed for mobility
Physiotherapy is important to prevent joint deformities and to improve functional performances after the adoption of appropriate shoes and orthopaedic supports.
Orthopaedic surgery may be useful both for preventing or correcting joint deformities (as needed for severe pes cavus) and to stabilise joints which are no longer properly supported by muscles.
Drugs, such as acetaminophen or nonsteroidal anti-inflammatory agents for musculoskeletal pain and tricyclic antidepressants or carbamazepine or gabapentin for neuropathic pain, may be useful, too.
Psychological support is useful to establish a pathway of help and acceptance of the disease and for career/employment counseling.
Gait, strength, and visual acuity are to be kept under surveillance by annual evaluation.
Circumstances to avoid are obesity (which makes ambulation more difficult); medications (e.g., vincristine, isoniazid, nitrofurantoin) known to cause nerve damage; alcohol and malnutrition, which can cause or exacerbate neuropathy.
(Experimental therapeutics in hereditary neuropathies: the past, the present, and the future, 2008).
Authors
Martina Mazzoli
Francesca Toja