The Parkinson’s Disease (PD) is a progressive neurodegenerative disorder characterized by bradykinesia, resting tremor, rigidity and postural instability. It is caused by an irreversible degeneration of dopaminergic neurons in the pars compacta region of substantia nigra.
The loss of these neurons leads to alterations in the activity of the neural circuits within the basal ganglia that regulate movements, resulting in movement disorders.
PD is also characterized by the presence of intracytoplasmic inclusions known as Lewy Bodies into dopaminergic neuron. Lewy’s Bodies are made of alpha-synuclein , which is involved in PD pathogenesis as seen in case of mutation of its gene SNCA that leads to familial cases of PD.
Pathogenesis
This disease has been considered to be idiopathic for long time, but actually it has been shown that etiopathogenesis includes genetic, epigenetic and environmental factors. it is known that several mechanisms are involved in the development of PD pathogenesis such as mitochondrial dysfunction, oxidative damage, autophagic alterations, proteasome impairment, microtubule network disruption, and protein aggregation.
Nowadays mitochondrial dysfunction has been demonstrated as one of the leading event in PD pathogenesis.
This link was seen for the first time in drug abusers who accidentally exposed to 1-methyl-4-phenyl-1,2,3,4-tetrahydropyridine (MPTP) and manifested symptoms of Parkinson’s.
MPTP is an inhibitor of complex I of the mitochondrial respiratory chain and MPTP deficiency has been seen also in other tissue in PD patient. (Mitochondrial Dysfunction: The Road to Alpha-Synuclein Oligomerization in PD, 2011)
More evidences link mitochondrial dysfunction to PD and it has been suggested that dysfunctions occur prior to the symptoms of disease.
In facts, mitochondria play a vital role in neuronal cell survival: they regulate energy metabolism as ATP producers, homeostasis of intracellular calcium, NAD+/NADH ratio, production of endogenous ROS and they also participate in cell apoptosis program via c cytocrome pathway. ROS production is thought to be the main cause of mitochondrial dysfunction, which leads to misfolding of Alpha-synuclein and to cell death.
So what protects mitochondria from oxidative damage?
Several studies conducted on PD and other neurodegenerative disease patient have demonstrated that the main antioxidant lacking in these patient, especially PD ones, was the Coenzyme Q 10, an important component of respiratory chain that regulates also oxidative stress in cell.

Pathophisiology of PD: the role of mythocondrial disfunction Co-Q10 biochemistry and deficiency
Biochemistry
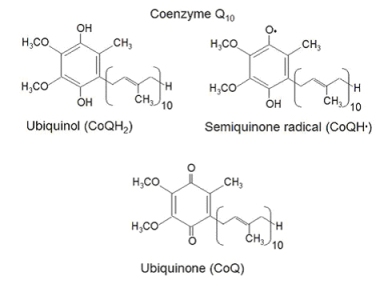
The Coenzyme Q-10 , or CoQ10 (2,3-dimethoxy, 5-methyl, 6-polyisoprene para-benzoquinone) or ubiquinone 50 is a lipophilic, redox active molecule located in all cellular membranes. It is composed by a quinone ring, that forms the head, and a hydrofobic tail, which contains 10 isoprenoid units, as the name itself says.
Ubiquinone is present in all tissue, but the highest concentrations are found in heart, brain, kidney and liver. CoQ10 is naturally present in vegetables (oil, beans, spinach, soya seed)
Coenzyme Q-10 presents in 2 different forms: the oxidized one, ubiquinone, and the reduced one, ubiquinol. Ubiquinol can be recycled from ubiquinone by CoQ10 reductases, such as NADH/quinone reductase and NADH/cytochrome b5 reductase.
Ubiquinone can accept one or two electrons both to transfer them into the respiratory chain, from complex I and II to complex III for ATP production, and to protect cell from oxidative stress.
When accepting one electron, it forms a semiquinone radical that is very reactive and can accept another electron to become the reduced form ubiquinol.
Biosynthesis of Coenzyme Q10
Nine genes are presumed to be involved in CoQ10 biosynthesis, and actually mutations in three genes have been shown to result in primary CoQ10 deficiency: PDSS1, PDSS2, and COQ2. (Coenzyme Q10 effects in neurodegenerative disease)
Ubiquinone is totally synthesized in mitochondrial matrix in a very complex way.
The first step of CoQ10 synthesis is the polyisoprenoid tail assembling by a polyprenyl diphosphate synthase. Next, COQ2 (4 hydroxybenzoate polyprenyltransferase) catalyses the formation of the covalent linkage between the benzoquinone head group and the tail to produce 4 hydroxy 3 polyprenyl benzoic acid intermediate (DHB, 3 decaprenyl 4 hydroxybenzoic acid in humans).
Then follows modifications of the aromatic ring starting with hydroxylation, followed by O methylation, and decarboxylation to form the 2 methoxy 6 polyprenyl phenol intermediate (DMPhOH ; 2 methoxy 6 decaprenylphenol in humans). Following this, two additional methylations, one C methylation, and one O methylation step, finally generate the fully substituted hydroquinone, ubiquinol.
Functions of CoQ10
This enzyme is an essential component of mitochondrial respiratory chain, where it transfers electrons from complex I and II to complex III to produce ATP and it is an inhibitor of the mitochondrial permeability transition pore in the inner mitochondrial

CoQ10 is implied in redox reactions in other cell membranes, such as Golgi vesicles, lysosomes, and plasma membrane.
This is a powerful antioxidant that protects cell from ROS-induced damage in two ways: directly reacting with ROS and regenerating alpha-tocopherol and ascorbate.
The reduced form, ubiquinol, is a powerful antioxidant that prevents oxidative damage by free radicals, including oxidation of lipids within the mitochondrial membrane. CoQ10 also serves as an antioxidant by activating and increasing expression of mitochondrial uncoupling proteins (UCPs), an effect which is antiapoptotic and leads to a reduction in free radical generation.
Moreover, CoQ10 has neuroprotective effects by increases brain mitochondrial functions
Deficiency of CoQ10 and mithocondrial disfunction in PD
Reduced CoQ10 levels, a tipical feature of PD patients, lead to mitochondrial energy production deficit. In humans, some neurodegenerative diseases are related to deficiency of this antioxidant enzyme. In Parkinson’s disease there are some evidences of abnormal mitochondrial function and excessive ROS e RNS damage which are responsible of the pathophysiology of this disease.
Free radical levels are significantly increased in cells with depleted CoQ10 levels.
The CoQ10 deficiency in the respiratory chain provides an excess of radical oxygen and nitrogen species due to the lack of its scavenging functions.
The ubiquinon disfunctions is one of the multiple mithocondrial alterations in the cell. Various defects in the complex of respiratory chain can lead to the loss of normal transfer of electrons and protons across the mitochondrial membranes.
Moreover, aging seems to affect mitochondria particularly. Mitochondria undergo the most dramatic age-related changes, such as structural deterioration like swelling and loss of cristae, sometimes complete destruction of the inner membrane, decreased respiration, and low ATP production.
Mt-DNA alterations may lead to a massive mitochondrial impairment triggering the death of dopaminergic neurons. When mitochondrial dysfunction reaches a critical threshold inside the cell it rouses ATP depletion, oxidative stress, and calcium homeostasis deregulation that in turn induces microtubule disruption, and alpha-synuclein oxidation.
Actually, very high levels of mtDNA deletions have been observed in dopaminergic neurons from the substantia nigra of postmortem human brains from aged individuals and idiopathic PD patients .
mtDNA deletions in these neurons were correlated with a decrease in Cytochrome C Oxidase activity, one of the key enzymes of electron transport chain. This suggests that accumulation of mtDNA deletions in dopaminergic neurons from substantia nigra above a certain threshold can cause mitochondrial defects associated with PD (Mitochondrial Dysfunction: The Road to Alpha-Synuclein Oligomerization in PD, 2011)
Nonfunctional mitochondria are important sources of ROS which contribute to the accumulation of damaged in domaminergic neurons. In addition, because of mitochondrial ROS generation, protein damage occurs and mtDNA mutations accumulate at an accelerating rate: this leads to the synthesis of abnormal mitochondrial proteins exacerbating mitochondrial dysfunction.
The role of ROS in alpha-synuclein olygomerization
As written above, the main pathological moment is the overproduction of ROS (reactive oxygen species) into mitochondria that subsequently leads to cell death.
ROS are a family of extremely reactive molecules which have an unpaired electron that react with other substances.
The three main component of this family are superoxide (O 2 -), hydrogen peroxide (H 2 O 2 ) and hydroxyl radical (·HO).
All of them are produced in mitochondria during oxidative phosphorylation and can potentially damage various type of biomolecules, including proteins, lipids, and nucleic acids.
Markers of oxidation are increased in substantia nigra samples of PD patient, making a relationship between this kind of damage and the onset of disease.
The main compound related to cytotoxicity in dopaminergic neuronal cell is H 2 O 2 .
Recent studies have revealed that nitration and nitrosylation of protein such as alpha synuclein is very related to PD.
Alpha synuclein is a small protein made of 140 amino acids, coded by SNCA gene, mainly expressed in CNS, in neocortex, thalamus, hippocampus, substantia nigra and cerebellum. We don’t know much about its function, but it’s hypothesized a role in the biosynthetic pathway of dopamine as an activator of protein phosphatase 2A and reducing levels of MAP-Kinase and phospholipase D in neuronal cell. Another suggested function of this protein is modulation of synaptic transmission, synaptic vesicles density and neuronal plasticity.
The damage to neuronal cells comes from the aggregation of alpha-synuclein monomers into alpha-synuclein oligomers. The oligomerization of this protein is mediated by interaction with calpain-1, that promotes the formation of fibrils, as also the interaction between alpha synuclein and alpha-tubulin does.
Moreover, alpha-synuclein is present in the membrane of mitochondria in normal dopaminergic neurons but, under overexpression conditions, it may translocate into the matrix and cause enhanced toxicity in response to subtoxic concentrations of mitochondrial toxins.

One promoter alfa synuclein aggregation is the mitochondrial-driven oxidative stress that enhances the stability of oligomers and facilitates aggregation. Another cause of the formation of oligomers is missense mutation in SNCA gene: A53T and A30P are known to be associated to rapidly self-aggregating form of alpha-synuclein. Only the first one A53T has shown development of neuronal synucleinopathy in mice. (Mitochondrial Dysfunction: The Road to Alpha-Synuclein Oligomerization in PD, 2011)
Oxidative stress, joined to the evidence that Lewy’s Bodies in dopaminergic neuronal cells are made of alpha-synuclein aggregates, represents the main pathogenetic event in development and progression of Parkinson’s Disease.

Administration Of CoQ-10 in PD patients
Since mitochondrial dysfunction and alpha-synuclein oxidation are both related to oxidative stress, that come from a deficient antioxidant system, in the pathogenesis of PD, one of the newest therapeutical approach to this progressive disease has been the administration of antioxidant therapy based on Coenzyme Q-10.
Actually, it is the CoQ10 deficiecy that provides an excess of ROS and RNS, so that the supplementation of this enzime can be usefull in PD patients.
Multiple in-vitro studies showed the neuroprotective effects of CoQ-10 in mouse samples, which decreases MPP-induced inhibition of complex I and II, tyrosine hydroxylases and hexokinase. It also reduces dopaminergic neuron iron-inducted apoptosis and rotenone-induced mitochondrial disfunction.
Administration of CoQ10 preserves mitochondria and decreases ROS production.
There are some evidences in literature that show the efficacy of CoQ10 treatment in PD patients:
Several studies has looked for an answer if the oral supplementation of CoQ-10 could be used as an efficient therapy to slow down the progression of PD. What some studies suggest is that administration of CoQ10 reduces the MPTP-induced neuronal cell damage.
A phase II clinical trial identified the optimal dose of therapy at 1200 mg/kg/day of CoQ10. Therapeutic effects of coenzyme Q10 (CoQ10) and reduced CoQ10 in the MPTP model of Parkinsonism, 2008
Conclusion
This represents a very important step forward in our knowledge of this disease and the results provide evidence that administration of CoQ10 may be a useful therapeutic strategy for the treatment of PD.
Mirko Domenico Iozzia
Martina Callegari