DEFINITION
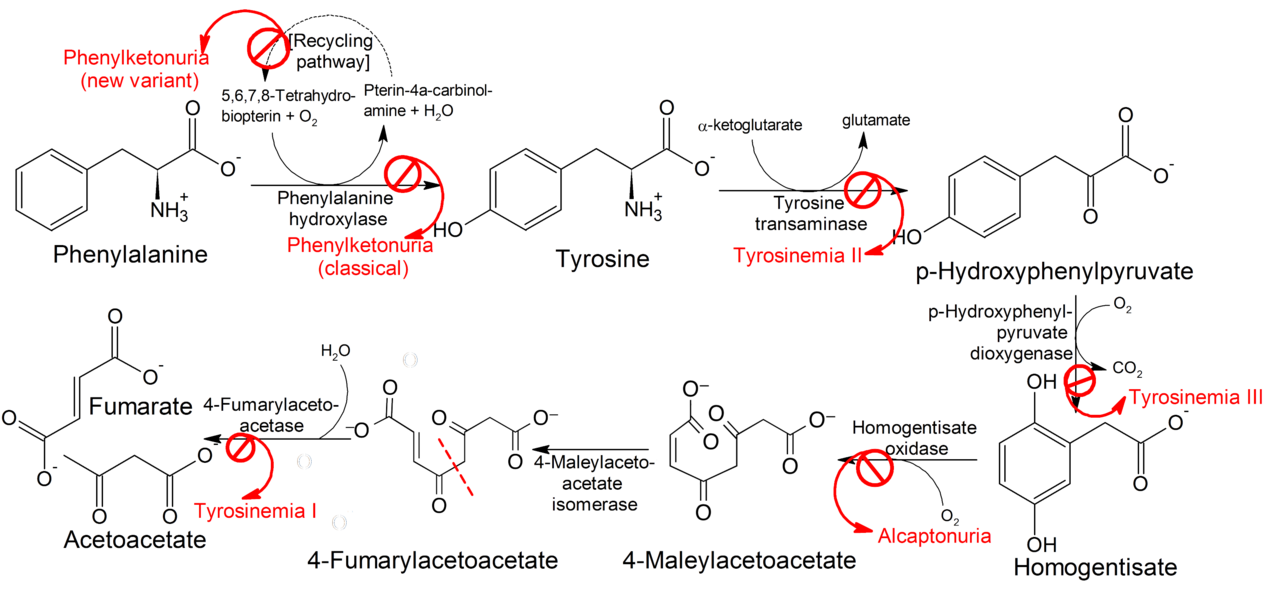
Tyrosinemia is a genetic disorder caused by the deficiency of one of the enzymes required for the multistep process that breaks down tyrosine. If untreated, tyrosine and its byproducts build up in tissues and organs, which leads to serious medical problems.
There are three types of tyrosinemia. Each has distinctive symptoms and is caused by the deficiency of a different enzyme:
Type I tyrosinemia, the most severe form of this disorder, is caused by a shortage of the enzyme fumarylacetoacetate hydrolase. It is also known as hereditary tyrosinemia, hepatorenal tyrosinemia, FAH deficiency, tyrosinosis and hypertyrosinemia
Type II tyrosinemia is caused by a deficiency of the enzyme tyrosine aminotransferase. This form of the disorder can affect the eyes, skin, and mental development.
Type III tyrosinemia is a rare disorder caused by a deficiency of the enzyme 4-hydroxyphenylpyruvate dioxygenase. Characteristic features include intellectual disability, seizures, and periodic loss of balance and coordination (intermittent ataxia).
About 10 percent of newborns have temporarily elevated levels of tyrosine, but, in these cases, the cause is not genetic. The most likely cause is immature liver enzymes due to premature birth and this condition is essentially benign and spontaneously disappears with no sequelae. Transient tyrosinemia is not categorized as an inborn error of metabolism because it is not caused by a genetic mutation.
Also the link to the corresponding Mesh term has to be created
EPIDEMIOLOGY
Tyrosinaemia I is much more common than type II; Type III is very rare.

Worldwide, type I tyrosinemia affects about 1 person in 100,000. This type of tyrosinemia is much more common in Quebec, Canada: the overall incidence in Quebec is about 1 in 16,000 individuals. In the Saguenay-Lac-Saint-Jean region of Quebec, type 1 tyrosinemia affects 1 person in 1,846. The carrier rate has been estimated to be between 1 in 20 and 1 in 31.
SYMPTOMS
Different babies may have different symptoms of tyrosinemia type I. Some may show symptoms within the first few months of life (acute form); others may develop symptoms around 1 year of age (chronic form). Some symptoms may be:
Acute form:
- poor appetite and failure to grow normally
- vomiting
- diarrhea, bloody stools
- a cabbage-like odor
- jaundice
- hepatomegaly
- lethargy
Chronic form:
- Cirrhosis of the liver
- Pain, numbness, and tingling in parts of the body (polyneuropathy; it developed because of the inhibitory effects of succinylacetone on the heme biosynthetic pathway)
- Kidney problems
- Episodes of intense abdominal pain
- Heart muscle weakness
Both forms of tyrosinemia type I may cause liver failure and/or develop liver cancer
DIAGNOSIS
Typical biochemical findings include:
- Increased succinylacetone concentration in the blood and urine;
- Elevated plasma concentrations of tyrosine, methionine, and phenylalanine;
- Elevated urinary concentration of tyrosine metabolites (p-hydroxyphenylpyruvate, p-hydroxyphenyllactate, and p-hydroxyphenylacetate)
- Increased urinary excretion of the compound δ-ALA (secondary to inhibition of the enzyme δ-ALA dehydratase by succinylacetone in the liver and circulating red blood cells)
- Decreased fumarylaceteoacetic acid hydrolase (FAH) enzyme activity (although
possible to assay in skin fibroblasts, this test is not readily available)
Tyrosinemia type I is typically detected on newborn screening and findings on newborn screening that suggest the diagnosis of tyrosinemia type I are:
- Elevated blood tyrosine or methionine concentration, that suggest liver disease),
- Succinylacetone, measured directly from the newborn blood spot by tandem mass spectroscopy
- Delta-ALA-dehydratase (PBG synthase) enzyme activity
Confirmation of diagnosis is usually by mutation analysis.
Prenatal diagnosis is feasible by mutation analysis on chorionic villus sampling (CVS), if the familial causative mutations are known or alternatively by FAH assay on CVS or amniocytes and determination of SA levels in amniotic fluid.
PATHOGENESIS
The biochemical basis for tyrosinemia type I remained enigmatic until the late 1970s, when researchers described a compound called succinylacetone found in the urine of infants with the condition.
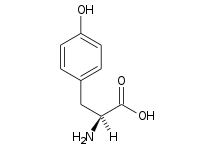
Succinylacetone was ultimately determined to be the decarboxylation product of succinyl acetoacetate, a compound derived from the tyrosine catabolic intermediate fumarylacetoacetate. Investigators inferred that the enzymatic defect might reside in deficiency of fumarylacetoacetase, which mediates production of fumaric acid and acetoacetate in both liver and kidney. This inference was later proven correct; succinyl acetoacetate accumulated because of this defect. Decarboxylation produced succinylacetone, which was then excreted in the urine, its source both hepatic and renal.
Although many aspects of the biochemical toxicity of this compound are known, the cellular basis for the multiorgan dysfunction found at the clinical level is unclear. In the kidney, succinylacetone has been demonstrated to be a mitochondrial toxin that inhibits substrate-level phosphorylation by means of the Krebs cycle. This compound also causes dysfunction of membrane transport in normal rat kidneys, altering membrane fluidity and possibly disrupting normal structure. It can cause renal tubular dysfunction in normal rat kidneys, mimicking human Fanconi syndrome, for which no other pharmacological animal model is available. Beyond its effects on the kidney, succinylacetone is a potent inhibitor of δ-aminolevulinic acid dehydratase, the enzyme that mediates formation of porphobilinogen, the cyclic precursor of porphyrins in the heme biosynthetic sequence. Succinylacetone-related alterations in heme biosynthesis of normal rat liver and kidney have been demonstrated. Recent data have suggested that fumarylacetoacetate itself induces mitotic abnormalities and instability in the genome. Research in murine animal models has indicated that this metabolite initiates apoptosis of hepatic and renal tubular cells. Taken together, these data form the basis for a unifying hypothesis regarding the development of hepatocellular carcinoma in children with hereditary tyrosinemia. A recent report indicates that fumarylacetocetate, but not succinylacetone, inhibits DNA glycosylases, which are instrumental in removing mutagenic base substitutions in the gene. Such data more firmly establish an etiologic relationship between the pathway intermediates and the mutational events they cause. Increased urinary excretion of δ-aminolevulinic acid can be attributed to inhibition of the heme biosynthetic pathway. A similar mechanism can account for the seizures commonly observed in patients; this mechanism is based on the demonstration of fumarylacetoacetase in the normal human brain. Absence of normal enzyme function could then be assumed to induce cellular accumulation of succinylacetone and to facilitate its toxic effects on the neuron.
Image source: Wikipedia
FAH gene

The FAH gene provides instructions for making the enzyme fumarylacetoacetate hydrolase. This enzyme is abundant in the liver and kidneys, and smaller amounts are found in many tissues throughout the body. Fumarylacetoacetate hydrolase is the last in a series of five enzymes needed to break down the amino acid tyrosine: specifically, fumarylacetoacetate hydrolase converts a tyrosine byproduct called fumarylacetoacetate into smaller molecules, fumarate and acetoacetate, that are either excreted by the kidneys or used in reactions that produce energy. Researchers have identified more than 40 FAH mutations that cause type I tyrosinemia. The altered FAH gene produces an unstable or inactive enzyme, which results in reduced or absent fumarylacetoacetate hydrolase activity.
The FAH gene is located on the long (q) arm of chromosome 15 at position 25.1 (Cytogenetic Location: 15q25.1 ; Molecular Location on chromosome 15: base pairs 80,152,890 to 80,186,581)
Image source: Genetics Home Reference
THERAPY
Nitisinone (Orfadin®). 2-(2-nitro-4-trifluoro-methylbenzyol) 1,3 cyclohexanedione (NTBC) was approved by the Food and Drug Administration in April 2002 for treatment of tyrosinemia type I. Nitisinone blocks parahydroxyphenylpyruvic acid dioxygenase (p-HPPD), the second step in the tyrosine degradation pathway, and prevents the accumulation of FAA and its conversion to succinylacetone. Nitisinone should be prescribed as soon as the diagnosis of tyrosinemia type I is confirmed.
Nitisinone increases blood concentration of tyrosine, necessitating a low-tyrosine diet to prevent tyrosine crystals from forming in the cornea. Dietary management should be started immediately upon diagnosis and should provide a nutritionally complete diet with controlled intakes of phenylalanine and tyrosine using a vegetarian diet with low-protein foods.
Prior to the availability of nitisinone for the treatment of tyrosinemia type I, the only definitive therapy was liver transplantation. Recent clinical experience indicates that liver transplantation should now be reserved for those children who have severe liver failure at clinical presentation and fail to respond to nitisinone therapy or have documented evidence of malignant changes in hepatic tissue. Transplant recipients require long-term immunosuppression and mortality associated with liver transplantation in young children is 10% or higher. Transplant recipients may also benefit from low-dose (0.1mg/kg/day) nitisinone therapy to prevent continued renal tubular and glomerular dysfunction resulting from persistence of succinylacetone in the plasma and urine.
Sonia Barello (Canale A)