NEONATAL HYPERAMMONEMIA
WHAT IS ORNITHINE TRANSCARBAMYLASE DEFICIENCY?
Ornithine transcarbamylase deficiency is an inherited disorder that causes ammonia to accumulate in the blood. Ammonia, which is formed when proteins are broken down in the body, is toxic if the levels become too high. The nervous system is especially sensitive to the effects of excess ammonia.
Ornithine transcarbamylase deficiency often becomes evident in the first few days of life. An infant with ornithine transcarbamylase deficiency may be lacking in energy (lethargic) or unwilling to eat, and have poorly-controlled breathing rate or body temperature. Some babies with this disorder may experience seizures or unusual body movements, or go into a coma. Complications from ornithine transcarbamylase deficiency may include developmental delay and intellectual disability. Progressive liver damage, skin lesions, and brittle hair may also be seen.
In some affected individuals, signs and symptoms of ornithine transcarbamylase may be less severe, and may not appear until later in life.
HOW COMMON IS ORNITHINE TRANSCARBAMYLASE DEFICIENCY?
Ornithine transcarbamylase deficiency is believed to occur in approximately 1 in every 80,000 people.
WHAT GENES ARE RELATED TO ORNITHINE TRANSCARBAMYLASE DEFICIENCY?
Mutations in the OTC gene cause ornithine transcarbamylase deficiency.
Ornithine transcarbamylase deficiency belongs to a class of genetic diseases called urea cycle disorders. The urea cycle is a sequence of reactions that occurs in liver cells. It processes excess nitrogen, generated when protein is used by the body, to make a compound called urea that is excreted by the kidneys.
Its gene can be regulated by glucocorticoids and other transcriptional factors such as C/EBP and HNF-4. The functional enzyme exists mostly as a trimer with an approximate molecular weight of 38 kDa. In humans OTC is localized in the mitochondrial matrix, mainly in the liver, but it is also in the intestinal epithelial cells. NcBi.
In ornithine transcarbamylase deficiency, the enzyme that starts a specific reaction within the urea cycle is damaged or missing. The urea cycle cannot proceed normally, and nitrogen accumulates in the bloodstream in the form of ammonia.
Ammonia is especially damaging to the nervous system, so ornithine transcarbamylase deficiency causes neurological problems as well as eventual damage to the liver.
HOW DO PEOPLE INHERIT ORNITHINE TRANSCARBAMYLASE DEFICIENCY?
Ornithine transcarbamylase deficiency is an X-linked disorder. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), mutations in both copies of the gene will cause the disorder. Some females with only one altered copy of the OTC gene also show signs and symptoms of ornithine transcarbamylase deficiency. GHR
WHAT IS THE NORMAL FUNCTION OF THE OTC GENE?
The OTC gene provides instructions for making the enzyme ornithine transcarbamylase.
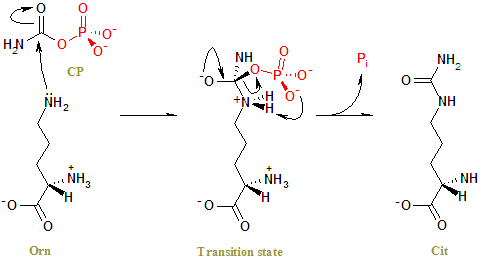
This enzyme participates in the urea cycle, a series of reactions that occurs in liver cells. The urea cycle processes excess nitrogen, generated when protein is used by the body, into a compound called urea that is excreted by the kidneys. Excreting the excess nitrogen prevents it from accumulating in the form of ammonia, which is toxic, especially to the nervous system.

The specific role of the ornithine transcarbamylase enzyme is to control the reaction in which two compounds, carbamoyl phosphate and ornithine, form a new compound called citrulline.
WHERE IS THE OTC GENE LOCATED?
Cytogenetic Location: Xp21.1
Molecular Location on the X chromosome: base pairs 38,352,482 to 38,421,449

The OTC gene is located on the short (p) arm of the X chromosome at position 21.1.
More precisely, the OTC gene is located from base pair 38,352,482 to base pair 38,421,449 on the X chromosome.
GHR
NEONATAL-ONSET OTC DEFICIENCY
Males with severe OTC deficiency are typically normal at birth, but become symptomatic on the second to third day of life with poor suck, reduced intake, and hypotonia, followed by lethargy progressing to somnolence and coma. They hyperventilate, and may have seizures. By the time neonates with OTC deficiency come to medical attention they typically are catastrophically ill with low body temperature (hypothermia), severe encephalopathy, and respiratory alkalosis.
When clinical and laboratory findings support the diagnosis of a urea cycle disorder, rescue therapy is begun immediately (see Management, Treatment of Manifestations).
The prognosis of a newborn in hyperammonemic coma depends on the duration of elevated ammonia level, not the height of the ammonia level or the presence or absence of seizures
Late-onset (partial) OTC deficiency
Hemizygous males and heterozygous females with partial OTC deficiency can present from infancy to later childhood, adolescence, or adulthood [Ahrens et al 1996, Ausems et al 1997, McCullough et al 2000]. Often they first become symptomatic in infancy when switched from breast milk to formula or whole milk (as breast milk contains less protein than infant formulas manufactured in the US). Infants may show episodic vomiting, lethargy, irritability, failure to thrive, and developmental delay. They show true protein avoidance, which can be documented by a detailed assessment of their dietary intake. When forced to eat high-protein content food, they may become symptomatic.
A stressor can cause an individual with partial OTC deficiency to become symptomatic at any age. In general the milder the disease, the later the onset and the stronger the stressor required to precipitate symptoms.
Adults with very mild disease have become symptomatic after crush injury, post-operative presentations [Chiong et al 2007, Hu et al 2007], when on a high protein diet [Ben-Ari et al 2010], during the post-partum period, during cancer therapy, and when treated with high dose corticosteroids [Lipskind et al 2011]. Treatment with valproate [Morgan et al 1987, Arn et al 1990, Honeycutt et al 1992] or haloperidol has been associated with hyperammonemic crises in persons with OTC deficiency [Rubenstein et al 1990, Leão 1995, Oechsner et al 1998, Thakur et al 2006].
When children, adolescents, or adults with late-onset disease become encephalopathic they may reach stage 2 coma [Plum & Posner 1982] with erratic behavior, combativeness, and delirium (e.g., not recognizing family members around them, unintelligible speech, etc.). They may come to medical attention if these behavioral abnormalities lead to an emergency medical or psychiatric evaluation.
Heterozygous Female
The phenotype of a heterozygous female can range from asymptomatic to significant symptoms with recurrent hyperammonemia and neurologic compromise depending on favorable vs. non-favorable X-chromosome inactivation ( Genetic Counseling, Related Genetic Counseling Issues). The risk for hyperammonemia in a heterozygous female is especially heightened in the postpartum period (see Management, Pregnancy Management).
Approximately 15% of heterozygous females are thought to become symptomatic during their lifetime [Batshaw et al 1986]; however, many heterozygous females exhibit mild symptoms, self-restrict protein intake, and are never diagnosed as being symptomatic; thus, the percent of symptomatic females may be higher.
PATHOPHYSIOLOGY
The ways in which an elevated ammonia level and its secondary effects such as a high glutamine level in brain may cause brain damage are unclear [Butterworth 2002]; however, its most severe acute consequence is cerebral edema.
The specific roles of ammonia in the development of cerebral edema are still under investigation but it may affect aquaporins and potassium channels and water and potassium homeostasis in brain [Lichter-Konecki 2008, Lichter-Konecki et al 2008, Albrecht et al 2010].
A role for increased glutamine synthesis in the development of brain edema has also long been postulated [Tanigami et al 2005, Zwingmann & Butterworth 2005]. High brain glutamine levels associated with low levels of myoinositol (a known brain organic osmolyte [Zwingmann et al 2004] was interpreted as evidence that the osmotic consequences of high glutamine levels are a cause of the brain damage in hyperammonemia. Desjardins et al [2012] recently described how trapping of glutamine inside the astrocyte, and possibly not increased synthesis, could lead to astrocyte swelling and encephalopathy. Finally, glutamate excitotoxicity is being discussed as a possible cause of the brain damage in hyperammonemia [Norenberg 1998].
Increased extracellular potassium levels (which would lower the seizure threshold) could be a possible explanation for the increased frequency of seizures during hyperammonemic coma or during the chronic mild hyperammonemia experienced by any person with a urea cycle disorder [Lichter-Konecki 2008, Lichter-Konecki et al 2008].
PLASMA AMMONIA CONCENTRATION
If the patient is encephalopathic, ammonia levels are typically above 200 μmol/L and often above 500-1000 μmol/L.
Note: The plasma ammonia concentration at which an individual becomes symptomatic varies but is generally above 100 μmol/L; in stage 2 coma [Plum & Posner 1982] the plasma concentration may be between 200 and 400 μmol/L; and in stage 3 to 4 coma, above 500 μmol/L. Of note, these levels are approximations and a wider range of elevated ammonia levels may be observed.
• If the plasma ammonia concentration is elevated, obtain:
On a newborn. Plasma amino acid (PAA) analysis; and inquire about the results of amino acid testing from the state NBS laboratory. In OTC deficiency, glutamine would be expected to be very high and citrulline to be very low.
INSTRUMENTAL EXAMS
Neurologic. The electroencephalogram (EEG) during hyperammonemic coma shows low voltage with slow waves and may include a burst suppression pattern in which the duration of the interburst interval correlates with the height of the ammonia levels.
Seizures are common during hyperammonemic coma and may only be detected on EEG. They do not indicate a poor prognosis. However, persons with urea cycle disorders may also be prone to having seizures independent of hyperammonemic episodes.
Neuroimaging studies reveal, during a crisis, cerebral edema with small ventricles, flattening of cerebral gyri, and low density of white matter.
Neonates who survived after prolonged coma had ventriculomegaly, diffuse brain atrophy (not affecting the cerebellum), low-density white matter defects, and injury to the bilateral lentiform nuclei and the deep sulci of the insular and perirolandic regions.
TREATMENT
Prevention of primary manifestations : If neonatal-onset OTC deficiency is diagnosed prenatally, intravenous (IV) treatment with ammonia scavengers within a few hours of birth (before the ammonia level rises) can prevent a hyperammonemic crisis and coma.
Prevention of secondary complications : Avoid over-restriction of protein/amino acids; use gastrostomy tube feedings as needed to help avoid malnutrition; practice careful hand hygiene among all that have contact with the patient to minimize risk of infection; give immunizations on the usual schedule, including annual flu vaccine; provide multivitamin and vitamin D supplementation; and use antipyretics appropriately (e.g., ibuprofen is preferred over acetaminophen because of the potential for liver toxicity).
Surveillance : At the start of therapy, routine measurement of plasma ammonia and plasma amino acids. Assess liver function (depending on symptoms) every three to six months or more often when previously abnormal. Perform neuropsychological testing at the time of expected significant developmental milestones.
Agents/circumstances to avoid: Valproate, haloperidol, systemic corticosteroids, fasting, and physical and psychological stress.
A rapid removal of ammonia by hemodialysis can decrease mortality and morbidity in the patients with severe increase of ammonia levels. However hemodialysis (HD) in infants and young children are technically difficult to perform. Continuous venovenous hemofiltration (CVVH) is increasingly used as an alternative for HD, but performing CVVH in a neonate can be problematic due to small body size and difficult vascular access. The authors reported a successful CVVH using umbilical vein as a vascular access site for ammonia removal in a neonate with OTC deficiency with progressive elevation of plasma ammonia. Technical problems, pitfalls in performing the CVVH, and how the authors overcame the problems are discussed. NCBI.
After successful rescue from neonatal hyperammonemic coma, infants with severe neonatal-onset OTC deficiency can easily become hyperammonemic again despite a low-protein diet and treatment with an oral ammonia scavenger. Even on maximum ammonia scavenger therapy a neonate with severe OTC deficiency may tolerate as little as 1.5 g/kg/day of protein (the minimum amount needed to grow), and growth may be along the third percentile for length.
After neonatal rescue therapy, a child with severe neonatal-onset disease can also experience a ‘honeymoon’ period in which the protein tolerance is so high, due to rapid growth, that the child is metabolically stable for some months before experiencing frequent hyperammonemic episodes.
Typically by age six months (if not sooner) a liver transplant is needed because of the effect of recurrent hyperammonemia on the brain and of prolonged hospitalizations on quality of life.
The overall outcome depends on the severity of brain damage during the initial hyperammonemic crisis and during subsequent hyperammonemic crises, as well as on the success of long-term treatment in maintaining metabolic balance and treating complications of the disease.
Long-term treatment (including restriction of protein intake, use of nitrogen scavengers, and in some cases liver transplantation) is aimed at promoting growth and development and preventing hyperammonemic episodes.