Definition

Progeria is an extremely rare genetic disease wherein symptoms resembling aspects of aging are manifested at an early age. The word “progeria” comes from the Greek words "pro" (πρό), meaning "before", and "géras" (γῆρας), meaning "old age". Progeria is an extremely rare genetic disease of childhood characterized by dramatic, premature aging. It affects children and gives them an appearance of accelerated aging. The classic type of Progeria (there are different forms) is Hutchinson-Gilford Progeria Syndrome (HGPS). This pathology was first described in an academic journal by Dr. Jonathan Hutchinson in 1886 and Dr. Hastings Gilford in 1897, both in England. Scientists are particularly interested in Progeria because it might reveal clues about the normal process of aging. For WHO International Classification of Diseases, Progeria causes endocrine, nutritional and metabolic diseases.
Epidemiology
Progeria is a rare condition that is remarkable because its symptoms strongly resemble normal human aging, but occur in young children. It usually is not passed down through families, in fact rarely is it seen in more than one child in a family.
At present there are 53 known cases of Progeria around the world and only 2 in the UK. There is a reported incidence of Progeria of approximately 1 in every 4 to 8 million newborns. Both boys and girls run an equal risk of having Progeria.
Progeria appears to affect children of all races equally. Over the last 15 years the following countries have had reported cases: Algeria, Argentina, Australia, Austria, Canada, China, Cuba, England, France, Germany, Israel, Italy, Mexico, the Netherlands, Poland, Puerto Rico, South Africa, South America, South Korea, Switzerland, Turkey, the US, Venezuela, Vietnam and Yugoslavia.
Sufferers have a very brief life expectancy and rarely live beyond 16. The oldest patient on record with this disease was 29 years old.
Causes

Progeria is caused by a mutation in a protein known as Lamin A, which results in a misshapen cell nucleus. Progeria is currently hypothesized to result from a gene mutation arising around the time of conception or shortly thereafter. The mechanism by which the misshapen nucleus leads to accelerated aging symptoms is not currently known.
Progeria is due to a single-letter "misspelling" in a gene on chromosome 1 that codes for lamin A, a protein that is a key component of the membrane surrounding the cell's nucleus. Most children with classic Progeria harbor exactly the same misspelling in the lamin A (LMNA) gene, a substitution of just a single DNA base (a change from cytosine C to thymine T) among the gene's 25,000 base pairs. In a few Progeria patients there may be a different single base substitution such as guanine (G) to adenine (A) just two bases upstream. In every instance, the parents are normal indicating that the misspelling is a new, or "de novo," mutation in the child. The minute change in the LMNA gene changes the way in which the sequence is spliced by the cell's protein-making machinery. The end result is the production of an abnormal lamin A protein that is missing a stretch of 50 aminoacids near one of its ends.
Lamins are classified as either A or B type according to their primary sequence, expression pattern, and biochemical properties. B-type lamins are expressed in all cells during development and in adult animals, whereas A-type lamins are expressed in differentiated cells. The LMNA gene encodes three A-type lamins: lamin A (LA), lamin C, and lamin A delta-10. Lamin A contains a C-terminal CAAX box, which undergoes methyl esterification and farnesylation. In the process of LA maturation, the C-terminal 18 residues, which include the modified C-terminal cysteine, are removed in two specific cleavage steps. The most frequent LMNA mutation in Progeria syndrome is a nucleotide substitution at position 1824, C-to-T, resulting in a silent gly-to-gly mutation at codon 608 (G608G) within exon 11 of the LMNA gene. This predicts a deletion of 50 basepairs of prelamin A near the C terminus. Low levels of both the mutant mRNA and the mutant protein, LA delta-50, are expressed in fibroblasts derived from Hutchinson-Gilford progeria syndrome patients. In addition, HutchinsonGilford progeria syndrome cell nuclei frequently display irregular shapes.
Different mutations in other regions of the LMNA gene are responsible for a half-dozen other rare, genetic disorders. Those disorders are: Emery-Dreifuss muscular dystrophy type 2, limb girdle muscular dystrophy type 1B, Charcot-Marie-Tooth disorder type 2B1, the Dunnigan type of familial partial lipodystrophy, mandibuloacral dysplasia and a familial form of dilated cardiomyopathy.
There currently are no diagnostic tests or treatments for Progeria which remains relentlessly progressive and fatal. Although Hutchinson (1886) and Guilford (1904) did describe the disorder, it was recorded that on "March 19, 1754, died in Glamorganshire of mere old age and a gradual decay of nature at 17 years and 2 months, Hopkins Hopkins, the little Welshman.... He never weighed more than 17 pounds but for three years past no more than 12."
The ultimate structural instability of the nucleus is believed to cause the aging signs experienced by those diagnosed with this syndrome.
Research into the causes of Progeria is ongoing. The genetic abnormality that is believed to cause the rapid aging process was identified in 2003 as a common link shared by a majority of sufferers with the disease. Scientists are not entirely certain how the Lamin A protein relates to overall human aging.

(a) In wild-type cells, lamin A is farnesylated at its C-terminal CAAX box; cleavage of lamin A by ZMPSTE24 normally removes the farnesylated cysteine residue (top). (b) Mutations in ZMPSTE24 cause permanent farnesylation of lamin A, leading to aberrant nuclear morphology and causing progeria (middle). Permanent farnesylation is also observed in HGPS cells and occurs in mouse models of the disease in which ZMPSTE4 is mutated. © Farnesyl transferase inhibitors reduce lamin A farnesylation but induce an alternative prenylation pathway leading to lamin A geranylgeranylation. Varela et al.1 inhibit both farnesylation and geranylgeranylation by using a combination of statins and aminobisphosphonates; the treatment inhibits both forms of lamin A prenylation, restores nuclear morphology and eases disease-related symptoms.
Inheritance
Experts do not believe that Progeria is hereditary. They say it is due to a rare gene change which happens purely by chance. A non-twin sibling runs the same risk of having Progeria as any other child from another family. In about 1 in every 100 cases of HGPS the syndrome is passed down to the next generation within the same family.
Classical HGPS is an autosomal dominant disorder, each patient arising through a spontaneous mutation in LMNA, consistent with the increased mean parental age.
Non-classical progeria has been reported as an autosomal recessively inherited disorder, either because of parental consanguinity or because of recurrence in siblings.
LMNA mutations acting as an autosomal recessive trait without any heterozygote phenotype have been reported.
Though the genetic trait for Progeria is dominant, it is not inherited. Children who develop this mutation are not born to parents who have the abnormality. Researchers believe the mutated code that alters the nucleotide sequence may occur in the egg or in one single sperm immediately prior to conception.
Symptoms

Children are born with this disease appearing healthy, and begin to slow their normal growth rates between 18 and 24 months of age.
Their bodies gradually experience hair and body fat loss. The skin starts to take on an aged appearance as the syndrome advances, and their joints become more stiff, sometimes resulting in hip dislocation.
It is characterized by dwarfism, baldness, pinched nose, small face and small jaw relative to the head size, delayed tooth formation, aged-looking skin, diminution of fat beneath the skin, stiff joints, and premature arteriosclerosis,frail bone structure. Teeth are often slow to appear and may not appear at all. Children also experience limited growth and have characteristically small faces with pinched features.
Progeria also causes medical problems typically associated with the elderly, mostly cardiovascular in nature. Heart attack or stroke is the leading cause of death for those with the disorder. Interestingly, certain conditions common among the elderly, such as cancer and Alzheimer's, are not symptoms of Progeria.
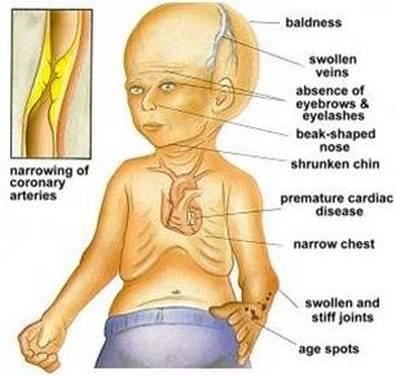
Then, Progeria's symptoms are:
• Growth failure during the first year of life
• Narrow, shrunken or wrinkled face
• Baldness
• Loss of eyebrows and eyelashes
• Short stature
• Large head for size of face (macrocephaly)
• Open soft spot (fontanelle)
• Small jaw (micrognathia)
• Dry, scaly, thin skin
• Limited range of motion
• Teeth - delayed or absent formation

Oral manifestation
Dental manifestations
-secondary incisors located lingually and palatally
-incomplete formation of roots of primary molars
-delayed tooth eruption of primary and secondary
dentition
-calcification along the nerve fibers and the vascular
walls
-abnormal tooth formation
-reticular atrophy of pulp
-anodontia
-delay in calcification of the crowns of the permanent
teeth
-hypodontia
-irregularity in calcification of the crowns of the
permanent teeth
-narrow pulp chambers
-discoloration
-high caries incidence
Head and Facial manifestations
-sparse to absent eyebrows and eyelashes
-delayed closure of fontanelles and sutures
-sparse to absent scalp hair
-no subcutaneous fat
-sculpted beaked nose
-relatively large tongue
-peri-oral cyanosis
-large cranium
-prominent forehead and frontal bossing
-small mouth
-prominent scalp veins
-prominent eyes
Jaw manifestations
-micrognathia
-comparative paucity of vertical growth
-hypoplastic mandible
-atrophy of alveolar process
-retarded anterior and vertical growth
-narrow and high palatal vault
-small maxillary arch
-short mandibular ramus
-obtuse mandibular angle
-craniofacial disproportion

The integral role of the dentist in treating individuals with Hutchinson-Gilford Progeria Syndrome
Degenerative Deseas
- Amyotrophic Lateral Sclerosis (ALS), or Lou Gehrig's Disease
- Alzheimer's Disease
- Parkinson's Disease
- Multiple system atrophy
- Niemann Pick disease
- Atherosclerosis
- Progressive supranuclear palsy
- Cancer
- Essential tremor
- Tay-Sachs Disease
- Diabetes
- Heart Disease
- Keratoconus
- Inflammatory Bowel Disease
- Prostatitis
- Osteoarthritis
- Osteoporosis
- Rheumatoid Arthritis
- Huntington's Disease
- Chronic traumatic encephalopathy
- Chronic Obstructive Pulmonary Disease
Syndromes
Hutchinson–Gilford progeria syndrome (HGPS) is a rare but well known entity characterized by extreme short stature, low body weight, early loss of hair, lipodystrophy, scleroderma, decreased joint mobility, osteolysis, and facial features that resemble aged persons. Cardiovascular compromise leads to early demise. Cognitive development is normal.
Data on 10 of our own cases and 132 cases from literature are presented. The incidence in the last century in the Netherlands was 1:4,000,000. Sex ratio was 1.2:1. Main first symptoms were failure to thrive (55%), hair loss (40%), skin problems (28%), and lipodystrophy (20%). Mean age at diagnosis was 2.9 years.
Growth in weight was more disturbed than growth in height, and growth delay started already prenatally. Mean height > 13 years was 109.0 cm, mean weight was 14.5 kg. Osteolysis was wide-spread but not expressed, except in the viscerocranium, and remained limited to membranous formed bone. Lipodystrophy is generalized, only intra-abdominal fat depositions remain present. Cardiovascular problems are extremely variable, both in age of onset and nature. Stroke and coronary dysfunctioning are most frequent. Pathologic findings in coronaries and aorta resemble sometimes the findings in elderly persons, but can also be much more limited. Loss of smooth muscle cells seems the most important finding. Mean age of demise was 12.6 years.
Patients can be subdivided in patients with classical HGPS, which follows an autosomal dominant pattern of inheritance, (almost) all cases representing spontaneous mutations, and in non-classical progeria, in whom growth can be less retarded, scalp hair remains present for a longer time, lipodystrophy is more slowly progressive, osteolysis is more expressed except in the face, and survival well into adulthood is not uncommon. Pattern of inheritance of non-classical progeria is most probably autosomal recessive.
The cause of HGPS is an abnormally formed Lamin A, either directly by a mutated LMNA gene, or through abnormal posttranslational processing (ZMPSTE24 gene mutations). Of 34 LMNA mutations found in Progeria patients, there were 26 classical p.G608G mutations (76%).
Pathogenesis is most likely to follow several different pathways. Potential therapeutic strategies are developed along these lines and include RNA interference techniques and inhibition of the dominant-negative influence of abnormally formed Lamin A on polymerization with normally formed Lamin A.
Cases of children with Syndrome of Hutchinson-Gilford
-Otlanmetse Falaste is the only black child diagnosed with Hutchinson-Gilford syndrome, she is 12 years old.

Otlanmetse Falaste's story I
Otlanmetse Falaste's story II
Otlanmetse Falaste's story III
- Dean Andrews is a Britain's oldest 20-year-old man has the body of a 160-year-old. He has aged eight times faster than normal due to Hutchinson-Gilford Progeria.

Dean Andrews's story
-Isabella Ceola is an Italian girl has written a book about herself, “io sono quella che sono... oggi posso dirvelo”, ed. Zanichelli. She died of old age at 28.

Isabella Ceola's story
-Hayley Okines is one of the first progeria children to try a new class of drug called FTIs (farnesyltransferase inhibitors).
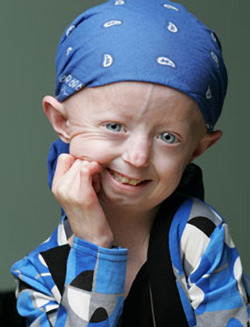
Hayley Okines's story I
Hayley Okines's story II
-Lindsay Ratcliffe, another child living with progeria.
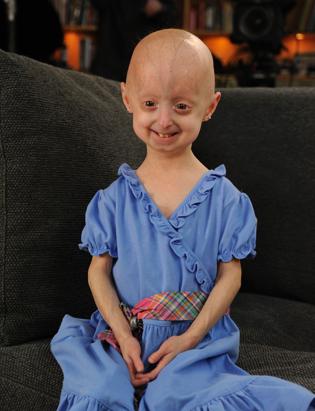
Lindsay Ratcliffe's story I
Lindsay Ratcliffe's story II
Werner syndrome (WS)

Werner syndrome is characterized by the dramatic, rapid appearance of features associated with normal aging. Individuals with this disorder typically grow and develop normally until they reach puberty. Affected teenagers usually do not have a growth spurt, resulting in short stature. The characteristic aged appearance of individuals with Werner syndrome typically begins to develop when they are in their twenties and includes graying and loss of hair; a hoarse voice; and thin, hardened skin. They may also have a facial appearance described as "bird-like." Many people with Werner syndrome have thin arms and legs and a thick trunk due to abnormal fat deposition.
As Werner syndrome progresses, affected individuals may develop disorders of aging early in life, such as cloudy lenses (cataracts) in both eyes, skin ulcers, type 2 diabetes, diminished fertility, severe hardening of the arteries (atherosclerosis), thinning of the bones (osteoporosis), and some types of cancer. It is not uncommon for affected individuals to develop multiple, rare cancers during their lifetime. People with Werner syndrome usually live into their late forties or early fifties. The most common causes of death are cancer and atherosclerosis.
Werner syndrome is estimated to affect 1 in 200,000 individuals in the United States. This syndrome occurs more often in Japan, affecting 1 in 20,000 to 1 in 40,000 people.
Mutations in the WRN gene cause Werner syndrome. The WRN gene provides instructions for producing the Werner protein, which is thought to perform several tasks related to the maintenance and repair of DNA. This protein also assists in the process of copying (replicating) DNA in preparation for cell division. Mutations in the WRN gene often lead to the production of an abnormally short, nonfunctional Werner protein. Research suggests that this shortened protein is not transported to the cell's nucleus, where it normally interacts with DNA. Evidence also suggests that the altered protein is broken down more quickly in the cell than the normal Werner protein. Researchers do not fully understand how WRN mutations cause the signs and symptoms of Werner syndrome. Cells with an altered Werner protein may divide more slowly or stop dividing earlier than normal, causing growth problems. Also, the altered protein may allow DNA damage to accumulate, which could impair normal cell activities and cause the health problems associated with this condition.
Werner syndrome is inherited in an autosomal recessive pattern, which means both copies of the WRN gene in each cell have mutations. The parents of an individual with Werner syndrome each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Neonatal Progeroid Syndrome
Neonatal progeroid syndrome is a premature aging syndrome in which features of human aging are apparent at birth, including larger than normal sized head; prominent scalp veins; triangular, aged face; wrinkled skin; and decreased fat under the skin. This differentiates this syndrome from other premature aging syndromes such as Hutchinson–Gilford Progeria syndrome (HGPS) (more commonly called Progeria) in which characteristics of premature aging typically become apparent sometime after birth. Although the exact cause of neonatal progeroid syndrome is unknown, it is believed to be genetic and inherited in an autosomal recessive fashion. Treatment is based on the individual's specific symptoms.
Hutchinson-Gilford progeria syndrome (HGPS) and mandibuloacral dysplasia are well-recognized allelic autosomal dominant and recessive progeroid disorders, respectively, due to mutations in lamin A/C (LMNA) gene. Heterozygous LMNA mutations have also been reported in a small number of patients with a less well-characterized atypical progeroid syndrome (APS).
A case of Atypical progeroid syndrome

Harry is aging at a rate that is five times faster than others his age, he is the only known person in the world with Atypical Progeria Syndrome.
Harry Crowther's story I
Harry Crowther's story II
Diagnosis
Doctors may suspect progeria based on signs and symptoms such as:
• Failure to grow
• Hair loss
To confirm the diagnosis, your doctor will order genetic testing. A genetic test for LMNA mutations can confirm the diagnosis of progeria.
The health care provider will perform a physical exam and order laboratory tests. This may show:
• Insulin resistance
• Skin changes similar to that seen in scleroderma (the connective tissue becomes tough and hardened)
Cardiac stress testing may reveal signs of early atherosclerosis of blood vessels.
Genetic testing can detect changes in the gene that causes progeria.
There are no specific tests to diagnose progeria. Identifying the disease often depends on inferences made from the physical appearance of the child within the first two years of birth, when changes begin to appear.
Recent research has revealed that elevated levels of hyaluronic acid can be detected in the urine of Progeria patients.
Therapy
It is important to note that there is no proven therapy as a cure for Progeria; however, there are theories that are based on clinical research that hope to further develop into possible cure.
Since Progeria patients have commonly low levels of antioxidants in the body, there are alternative therapies to combat oxidative stress.
Immediate Solutions:
- Prescribed formulas of antioxidants including vitamins, coenzyme Q-10, and lipoic acid
- Vitamin E for its antioxidant effects
- Treatment of Progeria with growth hormone
Oxygen levels are drastically impaired by high amounts of free radicals. Natural metabolic processes, toxins in the environment, and disease contribute to increased damage by radicals. Under normal conditions, enzymes called antioxidants break down free radicals and prevent the damage of body lipids, protein, and DNA. When free radicals supercede the amount of antioxidants in the body, the result is oxidative stress. Superoxide dismutase and catalase enzymes convert free radicals into benign substances such as water and oxygen. However, when these enzymes are found in small mounts, this can deplete the motor neurons of the oxygen they need to function. They are significant factors in determining the onset of the process of aging.
The nervous system is the most vulnerable system due to the amount of oxygen it consumes.
Long term solutions:
Genetic engineering can now be used to deliver genes that code for the enzymes into Progeria patients.
Progeria sufferers have only half of the normal caltase production of a normal metabolic percentage and only 30% of the glutathionine production required. Restoring their 80% deficient enzyme levels to normal could be the first step to combating the cause of this disease. If the right enzymes are produced it will halt the hyperdestruction of cells.
“Radical Medical Advancement” - The University of Washington is currently reintroducing enzymes that may halt or reverse the problem of age all together by using adenovirus gene transfers. By attaching enzymes to the common cold virus, the transporter (the cold virus) takes enzymes directly into the cell.
Once the telomeres reach a certain length on the chromosome, they cease to reproduce and the process of aging begins. A Progeria patient ages at a rate of seven times the normal aging process; therefore, targeting the area of actual rapid growth rate is of important consideration. Genetically defective telomeres could allow for cells to age faster and die faster than in a normal person. Telomeres are important because they protect chromosomes from degradation and fusion.
Since cellular aging is a genetically determined process, telomeres are the molecular clock for cellular aging. Manipulating the length of telomeres alters the life span of human cells.
When the gene that codes for the enzyme telomerase functions incorrectly they an build up at the end of genes and when the genes become shorter, they will stop dividing.
Human teleomerase composition - It is commonly found in immortal cells such as sperm. The catalytic component is hTERT enzyme.
New scientific findings belive that telomeres can now be cloned and then made into a drug that encourages cells to make their own telomerase.
Children with Progeria need Physical Therapy (PT) and Occupational Therapy (OT) as often as possible (optimally 2- 3 times each per week) to ensure maximum range of motion and optimal daily functioning throughout their lives.(07-04).pdf
Farnesyltransferase inhibitors (FTIs) might be useful in Hutchinson-Gilford Progeria Syndrome (2005)