DEFINITION
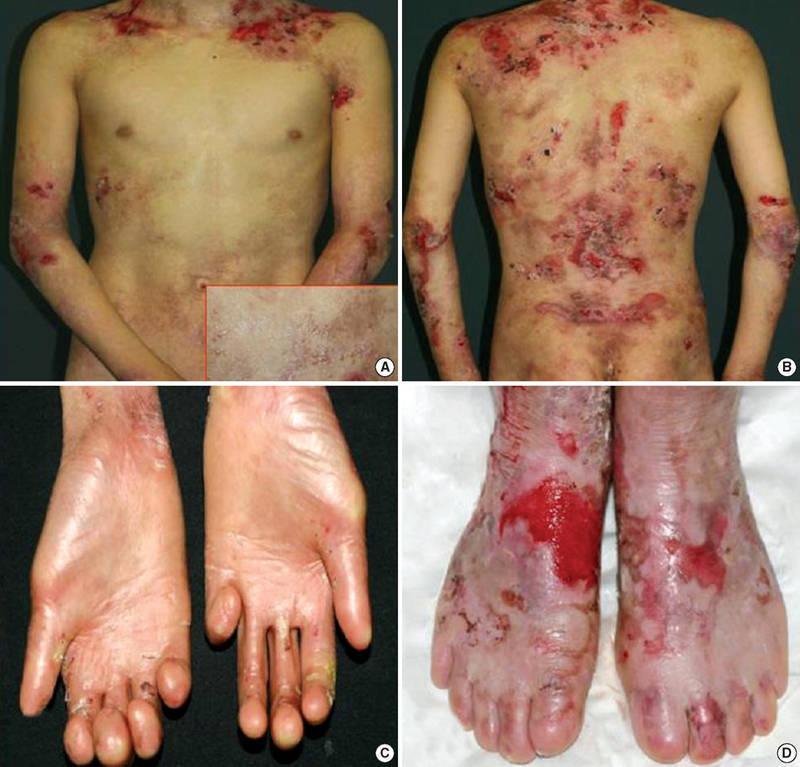
Epidermolysis bullosa is a group of genetic conditions that cause the skin to be very fragile and to blister easily. Blisters and skin erosions form in response to minor injury or friction, such as rubbing or scratching.
Dystrophic epidermolysis bullosa (DEB) is one of the major forms of epidermolysis bullosa. The signs and symptoms of this condition vary widely among affected individuals. In mild cases, blistering may primarily affect the hands, feet, knees, and elbows. Severe cases of this condition involve widespread blistering that can lead to vision loss, disfigurement, and other serious medical problems.
Wikipedia
Pubmed
OMIM
EPIDEMIOLOGY
There are three major types of DEB, all caused by mutations in the same gene (COL7A1). Considered together, the incidence of all types of dystrophic epidermolysis bullosa is estimated to be 6.5 per million newborns in the United States. The severe autosomal recessive forms (Hallopeau-Siemens type)of this disorder affect fewer than 1 per million newborns.
SYMPTOMS
| PRIMARY SYMPTOMS | CONSEQUENCES |
|---|
| skin blisters | scarring and secondary infection |
| ankyloglossia and microstomia | impaired food intake |
| esophageal erosions and web | dysphagia and impaired food intake |
| anal erosions | constipation |
| corneal erosions | scarring and loss of vision |
| urethral erosions, strictures |
| dilated cardiomyopathy |
| greater risk of developing SCC |
The impaired food intake can determine:
* growth retardation
* absent or delayed puberty
* anemia
* osteopenia and osteoporosis
More detailed description of symptoms
DIAGNOSIS
If there is a family history of the condition, the diagnosis is usually straightforward.
Tests that are used to confirm the diagnosis:
* Genetic testing
* Skin biopsy (usually with immunofluorescent tests or electron microscopy)
* Special microscopic tests of skin samples
Other tests that may be done:
* Blood test for anemia
* Culture to check for bacterial infection if wounds are healing poorly
* Upper endoscopy or an upper GI series if there are swallowing or feeding difficulties
Growth curves will be carefully watched in an infant who has, or is believed to have epidermolysis bullosa.
If there are contractures, limb range of motion will be tested.
More detailed description of diagnosis
INHERITANCE
Dystrophic epidermolysis bullosa (DEB) is inherited in an autosomal dominant or autosomal recessive manner.
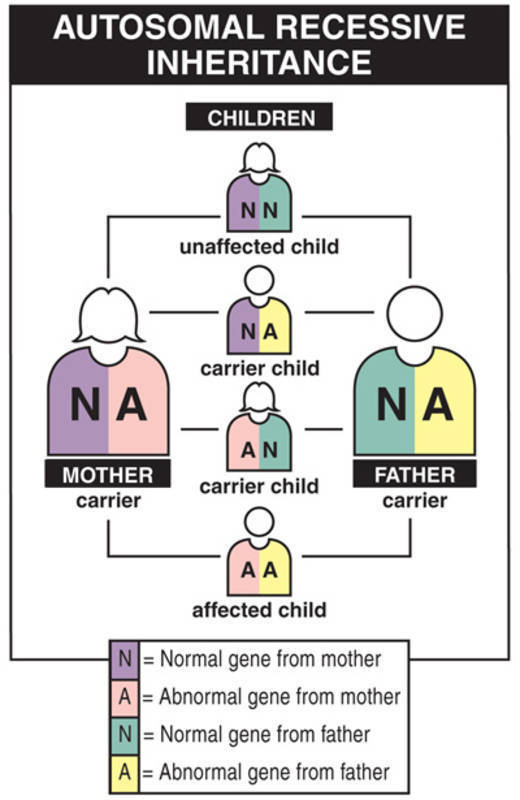
The most severe types of dystrophic epidermolysis bullosa are inherited in an autosomal recessive pattern. Autosomal recessive inheritance means that both copies of the COL7A1 gene in each cell have mutations. Most often, the parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but do not show signs and symptoms of the condition.
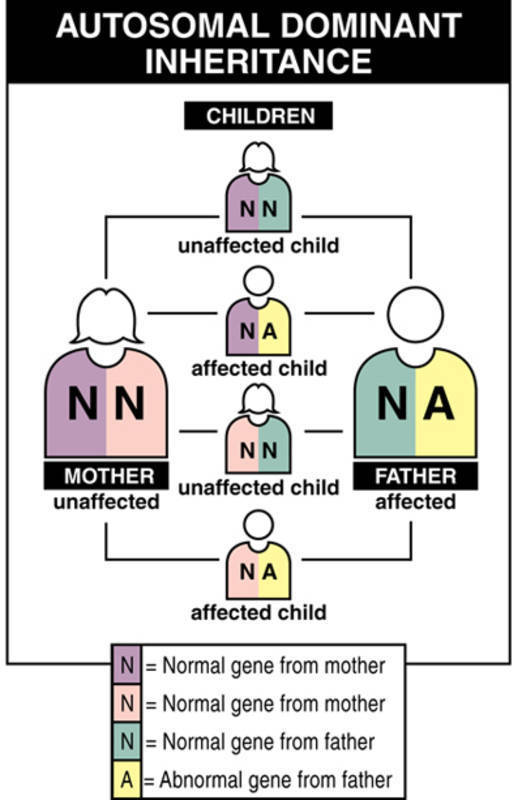
A milder form of dystrophic epidermolysis bullosa has an autosomal dominant pattern of inheritance. Autosomal dominant inheritance means that one copy of the altered gene in each cell is sufficient to cause the disorder. About 70 percent of all people with autosomal dominant dystrophic epidermolysis bullosa have inherited an altered COL7A1 gene from an affected parent. The remaining 30 percent of affected people have the condition as a result of a new mutation in the COL7A1 gene. These cases occur in people with no history of the disorder in their family.
PATHOGENESIS

Mutations in the COL7A1 gene cause all three major forms of dystrophic epidermolysis bullosa.
This gene provides instructions for making a protein that is used to assemble type VII collagen.
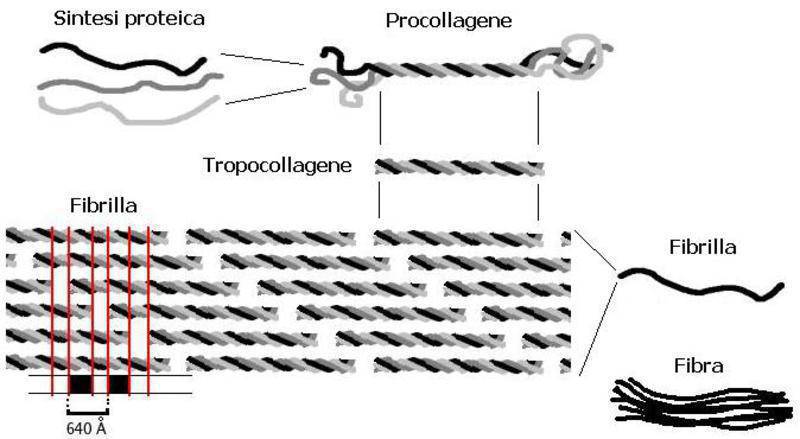
The COL7A1 gene is expressed by the basal keratinocytes of the epidermis where the protein products are assembled into homotrimeric molecules with a helical triple collagen domain. These molecules then assemble into homodimers that are found in the extracellular matrix below the lamina densa and form the anchoring fibrils that anchor the basement membrane to the underlying dermis.
The anchoring fibrils are linked to the basement membrane through attachment to laminin 5 and the keratinocyte hemidesmosomes directly above. These associations along with the network itself supply stability and resistance to stress that enable the keratinocytes to maintain their structural integrity during minor trauma and remain anchored to the basement membrane and dermis.

Mutations in the COL7A1 gene can lead to reduced resistance to minor trauma and the resulting blistering that is the hallmark of DEB. The type of mutation, the biochemical properties of the substituted amino acid, and its location determine the severity of the blistering phenotype and inheritance pattern.
* Missense mutations predominate in autosomal dominant forms of DEB and may affect the ability of the collagen VII to assemble into a triple helix (its secondary structure) and to form the intracellular network.
* Null mutations predominate in autosomal recessive forms of DEB and the absence of functional collagen VII and resulting absence of anchoring fibrils lead to the most severe forms of DEB.
Normal allelic variants : The normal cDNA comprises 9.2 kbp with an open reading frame of 8,833 nucleotides encoding 2944 amino acids in 118 exons spanning 32 kb.
Pathologic allelic variants : Glycine substitution mutations in the triple helical domain (especially in exons 73, 74, and 75) predominate (>75%) in DDEB. Mutations p.Gly2034Arg and p.Gly2043Arg are the most common DDEB-causing mutations, making up 50% of the dominant mutations reported in the largest US cohort.
Normal gene product : Collagen VII is a monomer of 2944 amino acids that associates into a homotrimer with a triple helical collagenous domain. The homotrimers then associate via disulfide bonds into homodimeric structures that form the anchoring fibrils.
Abnormal gene product : In dominant DEB, collagen VII with a glycine substitution in the collagenous domain may result in abnormal triple helical coiling and a partially nonfunctional protein product.
More information and genotype-phenotype correlations
THERAPY AND PROGNOSIS
Currently DEB cannot be cured. There are various treatments and surveillance methods for prevention of complications, many of them still under investigation.
Research is now focusing on gene replacement therapy.
Individuals with recessive DEB, formerly known as Hallopeau-Siemens type (RDEB-HS), have a greater-than-90% lifetime risk of aggressive metastasizing squamous cell carcinoma. The reason for the elevated risk has not been clear until recently. Ras-driven tumorigenesis in RDEB keratinocytes was examined and it was found that cells lacking collagen VII did not form tumors in mice, whereas those retaining a specific collagen VII fragment (the amino-terminal non-collagenous domain NC1) were tumorigenic. Restoring NC1 expression restored tumorigenicity in collagen VII-deficient cells. They conclude that tumor-stroma interactions mediated by collagen VII promote neoplasia, and retention of NC1 sequences in a subset of individuals with RDEB may be a factor in their increased susceptibility to squamous cell carcinoma.